Ion Adsorption in Porous Carbon Electrodes: Mechanisms, Materials, and Emerging Applications
This article provides a comprehensive examination of ion adsorption processes within porous carbon electrodes, a critical phenomenon for energy storage and environmental remediation technologies.
Ion Adsorption in Porous Carbon Electrodes: Mechanisms, Materials, and Emerging Applications
Abstract
This article provides a comprehensive examination of ion adsorption processes within porous carbon electrodes, a critical phenomenon for energy storage and environmental remediation technologies. It explores foundational principles, including entropy-driven design and solvation effects, and details methodological approaches for synthesizing and characterizing high-performance carbon materials. The content further addresses troubleshooting and optimization strategies for enhancing adsorption capacity and kinetics, and concludes with validation and comparative analyses of material performance. Tailored for researchers and scientists, this review synthesizes recent advances to guide the development of next-generation adsorbents for biomedical and clinical applications.
Unraveling the Core Principles of Ion Adsorption in Carbon Nanostructures
In the field of porous carbon electrodes for energy storage, the design of materials has traditionally focused on enthalpy-dominated strategies, such as maximizing surface area or introducing specific functional groups. However, a paradigm shift is emerging with the recognition of entropy as a critical, driving factor in adsorption processes and material stability. Entropy, a fundamental thermodynamic state function, measures the degree of disorder or randomness in a system. In adsorption phenomena, which are central to the performance of supercapacitors and batteries, there is always a decrease in the entropy (∆S < 0) of the adsorbate molecules as they become confined on the solid surface [1]. For the adsorption process to be spontaneous, this entropy loss must be compensated for by a sufficiently large, negative enthalpy change (∆H < 0), resulting in a net decrease in Gibbs free energy (∆G = ∆H – T∆S < 0) [1] [2].
The concept of "high-entropy materials" leverages configurational entropy—the entropy associated with the disordered mixing of multiple elements in a crystal lattice—to stabilize otherwise metastable structures and create surfaces with diverse adsorption sites [3] [4]. Recently, this design principle has been extended to carbonaceous systems, leading to the formal proposal of "high-entropy carbon materials" [3]. This novel class of materials is characterized by a high degree of structural disorder at the atomic scale, which can provide a variety of adsorption or reaction sites beneficial for electrochemical applications [3]. This technical guide explores the fundamental principles of entropy-driven adsorption, details the synthesis and characterization of high-entropy carbons, and situates these advancements within the broader context of ion adsorption research for next-generation porous carbon electrodes.
Core Principles of High-Entropy Carbon Materials
High-entropy carbon materials represent a departure from the ideal, crystalline graphene structure. Instead, they are designed around a "small graphene domain" model, where the carbon matrix is composed of numerous small, nano-sized graphene units that are highly disordered [3]. This inherent disorder is engineered through three distinct but complementary entropy-driven strategies, as summarized in the table below.
Table 1: The Three Design Principles of High-Entropy Carbon Materials
| Principle | Description | Mechanism of Entropy Increase | Key Structural Feature |
|---|---|---|---|
| Unit Entropy | Decreasing the size of graphene domains and increasing the number of basic structural units [3]. | Increases the system's configurational entropy by maximizing the number of possible arrangements for the small graphene domains [3]. | Disordered assembly of nano-graphene units. |
| Ring Entropy | Introducing topological defects that distort the ideal hexagonal carbon lattice [3]. | Replacing symmetric six-membered carbon rings with asymmetric five- and seven-membered rings increases topological disorder [3]. | 5-/7-membered carbon ring topological defects. |
| Element Entropy | Doping the carbon lattice with multiple non-metallic or metallic elements [3]. | The random incorporation of different heteroatoms (e.g., N, S, B, P) maximizes the configurational entropy of the chemical composition [3]. | Multi-element doping within the graphene lattice. |
The synergistic effect of these principles results in a carbon material with a high density of topological defects and heteroatoms. These features create a complex energy landscape with a variety of adsorption sites, which can lead to enhanced capacitive performance through both electric double-layer formation and pseudocapacitive interactions [3].
Quantitative Foundations: Entropy and Enthalpy in Adsorption
A quantitative understanding of adsorption thermodynamics is crucial for designing advanced porous carbon electrodes. The following table compiles key thermodynamic parameters from experimental studies on adsorption systems.
Table 2: Experimental Thermodynamic Parameters for Adsorption
| Adsorbate / System | Adsorbent | Enthalpy (ΔH) | Entropy (ΔS) | Experimental Method |
|---|---|---|---|---|
| n-Butane [5] | Activated Carbons | 49.1 - 53.4 kJ mol⁻¹ | ~116 J mol⁻¹ K⁻¹ | Adsorption Isotherms |
| General Physisorption [1] [2] | Solids | 20 - 40 kJ mol⁻¹ | Not Specified | Thermodynamic Analysis |
| General Chemisorption [1] [2] | Solids | 80 - 240 kJ mol⁻¹ | Not Specified | Thermodynamic Analysis |
| Adsorbed Molecules (at high coverage) [6] | Single Crystal Surfaces | Derived from Entropy | ( S{ad}^0(T) \approx 0.70 \ S{gas}^0(T) - 3.3R ) | Temperature Programmed Desorption (TPD) |
A landmark study on the entropies of adsorbed molecules revealed a robust linear correlation between the standard entropy of an adsorbate (( S{ad}^0 )) and the entropy of its gas-phase counterpart (( S{gas}^0 )): ( S{ad}^0(T) = 0.70 \ S{gas}^0(T) - 3.3R ), where R is the gas constant [6]. This relationship indicates that adsorbed molecules retain a significant portion (approximately two-thirds) of their gas-phase entropy, which is substantially higher than many theoretical predictions. This retained entropy is attributed to the preservation of various motional degrees of freedom upon surface confinement [6]. This finding is critical for accurately predicting reaction equilibria and rates in surface processes involving porous carbon electrodes.
Experimental and Computational Methodologies
Key Experimental Protocols
Research into entropy-driven adsorption and high-entropy materials relies on a suite of advanced characterization and computational techniques.
Temperature Programmed Desorption (TPD) for Entropy Determination
- Purpose: To experimentally determine the standard entropy (( S{ad}^0 )) and enthalpy (( H{ad}^0 )) of adsorbed molecules on well-defined surfaces [6].
- Procedure: A surface is saturated with the adsorbate at a low temperature. The temperature is then increased linearly while the partial pressure of the desorbing molecules is monitored with a mass spectrometer. The shape and position of the desorption peak are analyzed to extract kinetic and thermodynamic parameters [6].
- Data Analysis: The entropy can be calculated from the desorption activation energy and the pre-exponential factor in the Arrhenius rate constant. This method has been validated against equilibrium adsorption isotherms and provides reliable standard entropies for adsorbates [6].
Inverse Gas Chromatography (IGC)
- Purpose: To probe the surface properties and thermodynamic interactions of solid materials, such as porous carbons [5].
- Procedure: The solid material of interest is packed into a chromatography column. Known probe vapors are injected into the carrier gas stream at infinite dilution (to study high-energy sites) or finite concentration. The retention time of the probe molecules is measured [5].
- Data Analysis: The specific retention volume (( V_g )) is calculated from the retention time. This value is used to determine thermodynamic parameters, including adsorption enthalpy and entropy, dispersive surface energy, and surface heterogeneity [5].
Synthesis of High-Entropy Carbon Materials
- Principle: Employing entropy-driven strategies to create disordered carbon structures with small graphene domains [3].
- Unit Entropy Control: Utilizing precursors or activation methods that prevent the growth of large, crystalline graphene sheets, instead promoting a cross-linked network of small graphene units.
- Ring Entropy Control: Using high-energy treatments (e.g., plasma, high-temperature annealing) or chemical agents to create topological defects (5- and 7-membered rings) within the carbon lattice.
- Element Entropy Control: Co-doping the carbon matrix with multiple heteroatoms during synthesis using solid, liquid, or gas-phase precursors containing elements like N, B, S, and P [3].
The Role of Machine Learning and Computational Modeling
The vast compositional space of high-entropy materials makes their design a formidable challenge. Machine Learning (ML) has emerged as a powerful tool to navigate this complexity [7]. ML models can accelerate the discovery and optimization of high-entropy carbon materials by:
- Predicting Properties: Mapping the relationship between composition, processing parameters, and functional properties like specific capacity or cycling stability [7] [4].
- Screening Candidates: Rapidly identifying promising multi-element compositions from thousands of possibilities, thus reducing reliance on trial-and-error experimentation [7].
- Uncovering Design Rules: Analyzing large datasets to reveal hidden patterns and principles that govern material behavior [7].
Furthermore, microkinetic modeling, often informed by data from Density Functional Theory (DFT) calculations, is used to build detailed kinetic models of surface reactions, such as propane dehydrogenation [5]. These models incorporate entropy and enthalpy values for each elementary step to predict overall reaction rates and dominant pathways, providing atomic-level insight into processes relevant to catalytic and energy storage applications [5].
Research and Optimization Workflow for High-Entropy Carbon Materials
The Scientist's Toolkit: Essential Reagents and Materials
The experimental research into high-entropy carbons and adsorption thermodynamics involves a range of specialized reagents and instruments.
Table 3: Key Research Reagents and Materials for High-Entropy Carbon Research
| Item / Technique | Function / Purpose | Specific Examples / Notes |
|---|---|---|
| Activated Charcoal / Porous Carbons | Model adsorbent for fundamental studies of physisorption and pore-size effects [5] [2]. | Coconut charcoal; used in n-butane adsorption studies [5] [2]. |
| Single Crystal Surfaces | Provides a well-defined, atomically flat surface for precise measurement of adsorbate entropy and enthalpy [6]. | Pt(111), Ni(111); used in Temperature Programmed Desorption (TPD) [6]. |
| Heteroatom Dopants | Introduces element entropy and creates specific adsorption/reaction sites in the carbon lattice [3]. | Nitrogen (N), Sulfur (S), Boron (B), Phosphorus (P) precursors. |
| Metallic Precursors | For synthesizing High-Entropy Alloys (HEAs) or doping carbon with multiple metal elements [4] [8]. | Salts of Pt, Fe, Co, Ni, Cu for HEA nanoparticles [8]. |
| Temperature Programmed Desorption (TPD) | Key technique for measuring the entropy and enthalpy of adsorbed species [6]. | Requires an ultra-high vacuum system, mass spectrometer, and controlled heating stage. |
| Inverse Gas Chromatography (IGC) | Characterizes surface energy, acidity/basicity, and other surface properties of porous carbons [5]. | Uses probe molecules like n-butane; calculates specific retention volume (Vg) [5]. |
The introduction of entropy as a primary design variable, culminating in the "high-entropy carbon" concept, marks a significant evolution in the field of porous carbon electrodes. By deliberately engineering disorder through unit, ring, and element entropy, researchers can create materials with a rich diversity of adsorption sites, potentially leading to superior capacitive performance and enhanced reaction kinetics [3]. This entropy-driven approach, combined with a deeper quantitative understanding of adsorbate entropies [6], provides a more complete thermodynamic framework for optimizing ion adsorption.
Future research will likely focus on the precise control and quantification of the different entropy contributions in carbon materials. The integration of machine learning will be indispensable for decoding the complex structure-property relationships in these disordered systems and for accelerating the design of next-generation high-entropy carbons [7] [4]. As these materials mature, they hold the potential to overcome the performance plateaus of traditional carbon electrodes, enabling advanced energy storage technologies that meet the growing demands for high power, long cycle life, and superior energy density.
The Role of Solvation and Ionophilicity/Ionophobicity in Spontaneous Physisorption
In the field of energy storage and water desalination technologies, such as supercapacitors and capacitive deionization (CDI), microporous carbon electrodes are widely used due to their high specific surface area and tunable pore structures [9]. The core functionality of these materials relies on the adsorption and electrosorption of ions. A critical, yet often overlooked, precursor to the application of an electric field is the phenomenon of spontaneous physisorption, where ions distribute themselves within the carbon micropores without an external driving force [10] [11]. This spontaneous distribution is governed by the intricate balance between ion-ion and ion-solvent interactions, and the interaction of these solvated ions with the carbon pore walls. The concepts of ionophilicity (pore affinity for ions) and ionophobicity (pore aversion to ions) have emerged as crucial descriptors for this behavior [10]. Understanding the role of solvation in determining the ionophilic or ionophobic character of a system is therefore fundamental to designing next-generation porous carbon electrodes for more efficient energy storage and desalination applications [9] [11].
Core Concepts and Definitions
The Solvation Shell and Its Energetics
Solvation refers to the organization of solvent molecules around a dissolved ion. In aqueous systems, water molecules, being highly polar, form a structured hydration shell around ions through strong ion-dipole interactions [12]. The strength and structure of this shell are ion-specific, depending on the ion's charge density, size, and polarity. The energy required to partially or completely remove this solvation shell—the solvation energy—is a key factor in adsorption processes. When an ion approaches the confined space of a carbon micropore, it must shed its solvation shell to a degree that depends on the pore diameter. This desolvation process has an associated energy penalty that significantly influences whether adsorption is spontaneous or requires external electrical work [10].
Ionophilicity vs. Ionophobicity
The terms ionophilicity and ionophobicity describe the inherent tendency of a microporous carbon system to spontaneously adsorb or repel electrolyte ions.
- Ionophilic Systems: Characterized by a spontaneous uptake of electrolyte into the carbon micropores. This occurs when the overall free energy change for physisorption is favorable, often when the ion-carbon interaction energy is sufficient to overcome the cost of partial desolvation [10] [11].
- Ionophobic Systems: Exhibit a spontaneous exclusion of electrolyte from the micropores. In these systems, the energy penalty associated with distorting or stripping the ion's solvation shell within the confined pore space dominates, making physisorption unfavorable without an applied potential [10].
Table 1: Key Characteristics of Ionophilic and Ionophobic Systems.
| Feature | Ionophilic System | Ionophobic System |
|---|---|---|
| Spontaneous Adsorption | High | Low or None |
| Primary Driving Force | Favorable ion-carbon interactions | Energy penalty for ion desolvation |
| Typical Solvation Energy | Lower (for the specific ion-carbon pair) | Higher |
| Impact on Electrosorption | Favors counter-ion adsorption [10] | Favors co-ion ejection [10] |
Experimental Investigation Using NMR Spectroscopy
Nuclear Magnetic Resonance (NMR) spectroscopy has proven to be a powerful tool for directly observing and quantifying ion behavior within microporous carbons, providing molecular-level insights that are difficult to obtain with other techniques [10] [11].
Key Experimental Protocol
The following methodology summarizes the experimental approach used in recent studies to investigate spontaneous physisorption [10] [11]:
- Material Preparation: Microporous carbon electrodes with well-characterized pore size distributions are prepared. Pore diameters are varied to study confinement effects.
- Electrolyte Selection: Aqueous electrolytes with ions of varying solvation energies are used, such as sodium sulfate (Na₂SO₄) and sodium bis(trifluoromethane)sulfonimide (NaTFSI).
- Spontaneous Loading: The carbon material is brought into contact with a controlled volume of electrolyte without an applied potential, allowing for spontaneous physisorption to reach equilibrium.
- Ex Situ NMR Measurement: Solid-state NMR is used to quantify the amount and environment of ions (e.g., ²³Na) that have partitioned into the micropores during spontaneous loading. This reveals the ionophilicity/ionophobicity of the system.
- In Situ NMR Measurement: A supercapacitor cell is constructed with the carbon electrodes and assembled inside an NMR spectrometer. The system is then studied under operating conditions (during charging/discharging) to correlate the spontaneous partitioning behavior with the operational charge-balancing mechanism.
Quantitative Findings from NMR Studies
Recent NMR investigations have yielded critical quantitative data on how solvation and pore size dictate adsorption behavior. The following table summarizes key findings from these studies.
Table 2: Summary of Quantitative Findings on Solvation and Adsorption from NMR Studies.
| Investigated Parameter | System A (Ionophilic) | System B (Ionophobic) | Measurement Technique |
|---|---|---|---|
| Example Electrolyte | Aqueous Na₂SO₄ [10] | Aqueous NaTFSI [10] | NMR Spectroscopy |
| Spontaneous Adsorption | High partitioning into micropores [10] | Low partitioning into micropores [10] | Quantification of adsorbate volume |
| Pore Size Effect | Micropore diameter influences partitioning and disturbs ion solvation [10] | Micropore diameter influences partitioning and disturbs ion solvation [10] | Pore-size dependent NMR measurements |
| Charge Mechanism | Prefers counter-ion adsorption [10] [11] | Prefers co-ion ejection [10] [11] | In situ NMR on a working supercapacitor |
| Hydration Shell Status | Confinement-driven changes to ion hydration [10] | Confinement-driven changes to ion hydration [10] | Analysis of NMR spectral shifts |
Diagram 1: Logical relationship from solvation to electrosorption mechanism.
The Researcher's Toolkit: Essential Materials and Reagents
This section details the key reagents and materials required to conduct research on solvation and spontaneous physisorption.
Table 3: Research Reagent Solutions and Essential Materials.
| Item Name | Function / Rationale | Example Specifications |
|---|---|---|
| Microporous Carbon | Core adsorbent material; pore size dictates confinement effects [10] [9]. | Tunable pore diameter (e.g., 0.8-2 nm), high specific surface area [10]. |
| Aqueous Na₂SO₄ Electrolyte | Represents an ionophilic system; smaller, highly hydrated ions [10]. | 0.1 M to 1 M concentration in deionized water [10]. |
| Aqueous NaTFSI Electrolyte | Represents an ionophobic system; bulkier, less hydrated anion [10]. | 0.1 M to 1 M concentration in deionized water [10]. |
| Nuclear Magnetic Resonance (NMR) Spectrometer | Directly quantifies ion adsorption and characterizes ion environment [10] [11]. | Solid-state capability, suitable nucleus (e.g., ²³Na) probe [11]. |
| Electrochemical Cell (for in situ NMR) | Allows for study of electrosorption mechanisms under operating conditions [10]. | NMR-compatible, two-electrode supercapacitor configuration [10] [11]. |
Advanced Material Engineering Strategies
Beyond understanding the fundamental mechanisms, recent research focuses on engineering carbon materials to optimize their interaction with ions.
- Defect Engineering: Introducing topological defects, such as five- or seven-membered carbon rings, into the graphene lattice can create localized sites with altered electronic properties and enhanced affinity for ions, effectively increasing ionophilicity [3].
- High-Entropy Carbon Design: A novel concept involves creating "high-entropy carbons" by incorporating multiple non-metallic or metallic elements into the carbon structure (element entropy) or by distorting the graphene lattice (ring entropy). This increases the diversity of adsorption sites, potentially leading to superior and more tunable adsorption properties [3].
- Sustainable Process Engineering: To address the environmental impact of traditional carbon synthesis, green activators (e.g., inorganic salts) and sustainable manufacturing processes are being developed to produce high-performance porous carbons without the use of highly corrosive or toxic chemicals [9].
Diagram 2: Experimental workflow for physisorption analysis.
The pursuit of high-performance porous carbon electrodes for energy storage has catalyzed a paradigm shift from purely structural design to atomic-level engineering. Within this domain, the concept of entropy—a fundamental thermodynamic property—has emerged as a powerful principle for tailoring the electrochemical properties of carbon materials [13]. High-entropy carbon materials are characterized by their configurational complexity, which arises from the strategic introduction of disorder across multiple atomic scales. This disorder enables precise tuning of electronic structures and surface reactivity, creating a diverse array of adsorption sites ideally suited for ion adsorption processes in supercapacitors and batteries [13] [14]. This technical guide delineates the three fundamental pillars of entropy-driven design in carbon materials—unit entropy, ring entropy, and element entropy—and establishes their critical role in enhancing ion adsorption capabilities for next-generation electrochemical systems.
Core Principles of Entropy in Carbon Materials
The design of high-entropy carbon materials operates on three distinct but interconnected principles, each manipulating atomic structure to increase configurational entropy and thereby enhance ion adsorption properties.
Unit Entropy
Unit entropy is achieved by reducing the size of ordered graphene domains within the carbon matrix while simultaneously increasing the total number of these basic structural units [13]. The "graphene domain" refers to localized regions of sp²-hybridized carbon atoms exhibiting crystalline order. From an ion adsorption perspective, this nanostructuring creates a higher density of edges and interfacial boundaries, which are electrochemically active sites that facilitate ion adsorption and charge transfer. The increased disorder at the unit level provides a greater diversity of adsorption environments for electrolyte ions, directly enhancing the charge storage capacity of electric double-layer capacitors (EDLCs) [13].
Ring Entropy
Ring entropy introduces topological defects through the controlled distortion of the ideal graphene-plane six-membered carbon rings, generating asymmetric five- and seven-membered carbon rings [13]. These topological defects create strain fields and local charge inhomogeneities that significantly alter the interaction energy between the carbon surface and approaching ions. For ion adsorption, these distorted rings function as preferential nucleation sites for ion coordination, effectively reducing the energy barrier for ion desolvation—a critical step in the formation of the electric double-layer within confined nanopores [13] [15].
Element Entropy
Element entropy incorporates multiple heteroatoms (both non-metallic and metallic) into the graphene lattice [13] [14]. This multi-element doping creates a complex energy landscape with varied adsorption sites, each exhibiting distinct binding affinities for different ion species. The synergistic effect of these heteroatoms modulates the local electronic structure and surface reactivity of the carbon material. For instance, experimental studies on biomass-derived carbon have demonstrated that doping with elements such as calcium (Ca) can dramatically increase adsorption energy for specific molecules and enhance charge transfer, thereby improving electrochemical response [14].
Table 1: Design Principles and Their Impacts on Ion Adsorption
| Entropy Principle | Structural Feature | Key Impact on Ion Adsorption |
|---|---|---|
| Unit Entropy | Small graphene domains, high domain count | Increases edge site density and interfacial boundaries for enhanced ion coordination [13] |
| Ring Entropy | 5-/7-membered carbon ring topological defects | Creates local strain and charge pockets that reduce ion desolvation energy barriers [13] |
| Element Entropy | Multi-element doping (e.g., O, N, S, P, Ca) | Provides diverse adsorption sites with tailored binding energies and charge transfer capabilities [13] [14] |
Experimental Protocols and Methodologies
Synthesis of High-Entropy Carbon Materials
Biomass-Derived Synthesis (Marine Waste Precursor) The utilization of marine biomass, such as Undaria pinnatifida, provides a sustainable route to intrinsically multi-element-doped high-entropy-like carbon materials [14].
- Pre-treatment: Wash the biomass with deionized water and hydrogen peroxide solution (H₂O₂, 30%) to remove impurities, followed by freeze-drying to preserve the innate hierarchical architecture [14].
- Carbonization: Subject the freeze-dried material to a step-wise thermal treatment in an inert atmosphere. For example, heat to 500°C with a defined heating rate and hold time. This process carbonizes the organic framework while retaining heteroatoms (e.g., Ca, P, S, Na) intrinsic to the biomass [14].
- Activation (Optional): Perform chemical or physical activation to further develop porosity. The resulting material exhibits a hierarchical turbinate-like porous structure and a self-doped multi-element composition [14].
General Principles for Entropy Introduction
- To Enhance Unit Entropy: Employ activation agents or severe thermal conditions that fragment large graphene domains, promoting the formation of numerous small, disordered units [13].
- To Enhance Ring Entropy: Control the carbonization temperature and time to favor the rearrangement of carbon bonds and the formation of non-hexagonal ring structures [13].
- To Enhance Element Entropy: Utilize precursors rich in multiple elements or employ post-synthesis doping methods (e.g., vapor deposition, solution impregnation) to introduce a variety of heteroatoms into the carbon lattice [14].
Characterization Techniques for Ion Adsorption Analysis
Thermogravimetric Analysis (TGA) TGA profiles the thermal stability and decomposition stages of the precursor material. The mass loss between 200-500°C is typically associated with the primary pyrolysis of volatile components, crucial for determining the optimal carbonization temperature [14].
Density Functional Theory (DFT) Calculations DFT is indispensable for quantifying the effect of entropy-driven modifications on ion adsorption energetics at the atomic level [14].
- Adsorption Energy Calculation: Models the interaction between an ion (or molecule) and the carbon surface. A more negative adsorption energy indicates a stronger, more favorable interaction. For instance, Ca doping in carbon models has been shown to increase the adsorption energy of N₂H₄ from -0.412 eV to -1.532 eV [14].
- Charge Transfer Analysis: Determines the number of electrons transferred (Δe) between the adsorbate and the carbon substrate upon adsorption. Enhanced charge transfer, such as the increase from 0.039e to 0.070e for N₂H₄ on a Ca-doped site, signifies improved electrochemical activity [14].
- Diffusion Barrier Assessment: Evaluates the energy barrier that ions must overcome to diffuse within the carbon pores, which is critical for the kinetics of charge/discharge cycles [15].
Electrochemical Validation
- Cyclic Voltammetry (CV) and Galvanostatic Charge-Discharge (GCD): Used to measure the specific capacitance, rate capability, and cycling stability of the material in a supercapacitor cell. A high specific capacitance (e.g., 273 F g⁻¹ reported for an optimized coal-derived PC) indicates effective ion adsorption [15].
Table 2: Key Parameters from Ion Adsorption Analysis via DFT
| Material System | Analyte | Adsorption Energy (eV) | Charge Transfer (Δe) | Key Finding |
|---|---|---|---|---|
| Baseline Carbon | N₂H₄ | -0.412 | 0.039 | Reference system with moderate interaction [14] |
| Ca-doped Carbon | N₂H₄ | -1.532 | 0.070 | Doping creates strong, electrochemically active sites [14] |
| Baseline Carbon | O₂ | -0.160 | 0.181 | Baseline for oxygen interaction [14] |
| Ca-doped Carbon | O₂ | -1.524 | 0.790 | Doping drastically enhances O₂ adsorption and charge transfer [14] |
The Scientist's Toolkit: Essential Research Reagents and Materials
The experimental pursuit of high-entropy carbon materials requires a specific set of precursors, reagents, and analytical tools.
Table 3: Essential Research Reagents and Materials
| Item Name | Function/Application | Representative Examples |
|---|---|---|
| Marine Biomass Precursor | Sustainable source of carbon and intrinsic multi-element dopants (Ca, P, S, Na) [14] | Undaria pinnatifida (seaweed) [14] |
| Chemical Activating Agents | Etch the carbon framework to create micropores and mesopores, enhancing unit entropy [13] | KOH, H₃PO₄, ZnCl₂ |
| Heteroatom Dopant Sources | Introduce foreign elements (e.g., N, S, B, P) into the carbon lattice to increase element entropy [13] [14] | Urea (N), Thiourea (S), Boric Acid (B) |
| Metal Salt Precursors | Source for metallic dopants (e.g., Ca) that modulate electronic structure and enhance surface reactivity [14] | Calcium salts (e.g., Ca(NO₃)₂) |
| Gas Sensing Analytes | Used to probe the surface reactivity and adsorption properties of the material [14] | Hydrazine (N₂H₄), Ammonia (NH₃), Oxygen (O₂) [14] |
| Computational Software | For DFT calculations to predict adsorption energies, charge transfer, and electronic properties [14] [15] | VASP, Quantum ESPRESSO, Gaussian |
Visualization of Concepts and Workflows
The following diagrams, generated with Graphviz, illustrate the core concepts and experimental workflows of entropy-driven design in carbon materials.
Three Pillars of High-Entropy Carbon
Ion Desolvation & Adsorption Mechanism
Biomass to Sensor Experimental Workflow
The atomic-level design of carbon materials through unit, ring, and element entropy represents a sophisticated approach to mastering ion adsorption phenomena. By deliberately engineering disorder across multiple structural hierarchies, researchers can create carbon electrodes with tailored pore geometries, optimized surface charge distributions, and enhanced affinities for specific ions. The principles outlined in this guide provide a foundational framework for developing next-generation energy storage systems, with applications ranging from high-performance supercapacitors to sensitive gas sensors [13] [14] [15]. Future research will likely focus on the precise quantitative mapping between specific entropy-enhancing modifications and their resultant electrochemical properties, further unlocking the potential of these complex, functional materials.
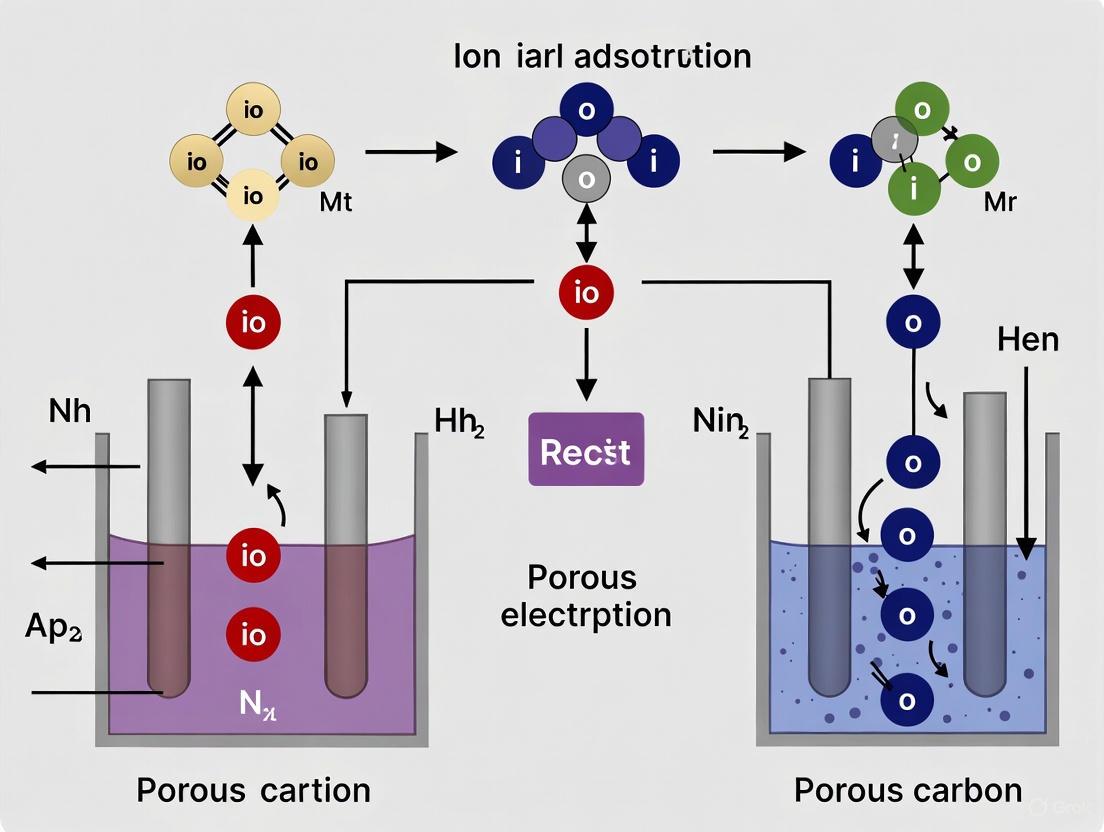
Ion electrosorption in porous carbon electrodes is a fundamental process underpinning technologies ranging from supercapacitors to capacitive deionization (CDI) for water treatment. The charge-balancing mechanism during electrosorption primarily occurs through two distinct pathways: counter-ion adsorption and co-ion ejection. The prevailing mechanism is not merely a theoretical distinction but has profound implications for the efficiency, capacity, and design of electrochemical systems. Recent advanced studies, particularly utilizing techniques like in situ nuclear magnetic resonance (NMR) spectroscopy, have demonstrated that the spontaneous ionophilicity or ionophobicity of the carbon-electrolyte system dictates which mechanism dominates [10]. This whitepaper provides an in-depth technical examination of these fundamental mechanisms, framed within the broader context of ion adsorption research for porous carbon electrodes, and equips researchers with the experimental and theoretical tools to characterize and optimize these processes.
Theoretical Foundations of Electrosorption
The Electrical Double Layer (EDL) in Confinement
At the heart of electrosorption lies the formation of an Electrical Double Layer (EDL) at the electrode-electrolyte interface. When a porous carbon electrode is polarized in an electrolyte, electronic charge accumulates in the carbon matrix. Electroneutrality requires this charge to be compensated by ionic charge in the adjacent electrolyte. The EDL is commonly described by the Gouy-Chapman-Stern model, which conceptualizes three key regions [16]:
- The Charged Carbon Matrix: Contains the electronic charge.
- The Stern Layer: A inner, compact layer of specifically adsorbed ions, separated from the carbon surface by a molecular dielectric layer.
- The Diffuse Layer: A region where ions are distributed diffusively, screening the electrode's charge. The characteristic thickness of this layer is approximated by the Debye length, which is about 3.1 nm for a 10 mM NaCl solution at 20°C [16].
In the confinement of carbon micropores (typically < 2 nm), the distinction between these layers can blur. The pore size becomes comparable to the Debye length and the solvation shells of ions, leading to significant distortions in ion solvation and unique ion packing, which dramatically influences the electrosorption mechanism [10] [15].
Defining the Core Mechanisms
The two primary charge-balancing mechanisms are defined by the type of ion that is mobilized to maintain electroneutrality upon electrode polarization.
- Counter-ion Adsorption: This mechanism involves the adsorption of ions with a charge opposite to that of the polarized electrode (e.g., cations moving to a negatively charged cathode). This leads to a net increase in the ion concentration within the electrode pore.
- Co-ion Ejection: This mechanism involves the expulsion (desorption) of ions with the same charge as the polarized electrode (e.g., cations being pushed out of a negatively charged cathode). This leads to a net decrease in the ion concentration within the pore.
The total salt adsorption is the net effect of these two competing processes. In Membrane Capacitive Deionization (MCDI), ion-exchange membranes are placed in front of the electrodes to block co-ion expulsion from the electrode, thereby enhancing the salt adsorption capacity and efficiency by forcing the system to rely predominantly on counter-ion adsorption [16].
Determinants of the Charge-Balancing Mechanism
The dominant mechanism in a given system is not arbitrary but is governed by the intrinsic properties of the electrode and electrolyte. In situ NMR studies have revealed that the spontaneous physisorption behavior of ions in the absence of an applied potential is a key predictor [10].
- Spontaneously Ionophilic Systems: In these systems, ions exhibit a thermodynamic preference to reside within the carbon micropores even at zero potential. When a potential is applied, these systems preferentially utilize counter-ion adsorption for charge compensation [10].
- Spontaneously Ionophobic Systems: In these systems, the carbon pores are naturally devoid of ions at zero potential. Upon polarization, these systems tend to favor the co-ion ejection mechanism to balance charge [10].
Several material and electrolyte properties determine this ionophilic/ionophobic character, as summarized in the table below.
Table 1: Key Factors Influencing the Dominant Electrosorption Mechanism
| Factor | Impact on Ionophilicity/Ionophobicity | Resulting Preference for Mechanism |
|---|---|---|
| Ion Solvation Energy [10] [15] | High solvation energy creates a large energy barrier for ion desolvation, favoring ionophobic behavior. Lower solvation energy promotes easier ion entry into pores (ionophilic). | High energy → Co-ion ejectionLow energy → Counter-ion adsorption |
| Pore Size / Diameter [10] [17] | Micropores (especially < 1 nm) can distort ion hydration shells. Optimal pore size matching ion diameter promotes ionophilicity. Mismatched pores are ionophobic. | Matched size → Counter-ion adsorptionMismatched → Co-ion ejection |
| Electrode Surface Chemistry [18] | Heteroatom doping (e.g., N, O) enhances surface polarity and specific ion interactions, generally promoting ionophilicity and specific counter-ion adsorption. | Doping → Counter-ion adsorption |
| Electrolyte Concentration | Higher concentrations compress the EDL (shorter Debye length), which can influence the relative contribution of each mechanism. | Complex, system-dependent |
Quantitative Data and Experimental Insights
Advanced characterization techniques have enabled the quantitative dissection of these mechanisms and their impact on performance.
Table 2: Quantitative Experimental Data on Ion Electrosorption Mechanisms
| Study System / Material | Key Measurement / Observation | Implication for Mechanism | Source |
|---|---|---|---|
| Microporous Carbon with Aqueous Na₂SO₄ & NaTFSI | In situ NMR revealed spontaneous ion partitioning. Ionophilic systems showed preference for counter-ion adsorption under potential. | Direct molecular-level evidence linking spontaneous adsorption to charge-balancing mechanism. | [10] |
| Porous Carbon for K⁺ storage | Identified 5 distinct desolvation states ([K(H₂O)₀₋₄]⁺). Quantified desolvation energies and diffusion barriers for each state. | High desolvation energy can limit counter-ion adsorption; kinetics and thermodynamics of desolvation are critical. | [15] |
| Biomass-derived Carbon for Zn²⁺ storage | Optimized pore diameter from 0.54 nm to 0.71/1.13 nm to match [Zn·(H₂O)₆]²⁺ (∼0.86 nm). Achieved high capacity of 269.54 mAh/g. | Pore size engineering to reduce desolvation penalty and promote efficient counter-ion adsorption. | [17] |
| N/O-codoped Dense Porous Carbon | DFT calculations confirmed enhanced K⁺ adsorption energy due to N/O doping. Achieved specific capacitance of 314 F/g. | Heteroatom doping thermodynamically favors counter-ion adsorption over co-ion ejection. | [18] |
Experimental and Computational Methodologies
Key Experimental Protocols
To investigate these mechanisms, researchers employ a suite of sophisticated techniques:
In situ Nuclear Magnetic Resonance (NMR) Spectroscopy
- Objective: To directly observe and quantify the partitioning and environment of ions within working micropores under potential control [10].
- Detailed Workflow: a. Cell Design: Fabricate an electrochemical NMR cell compatible with the spectrometer, ensuring the electrode material is packed appropriately. b. Isotopic Labelling: Use isotopes with favorable NMR properties (e.g., ²³Na) or deuterated solvents (D₂O) to minimize background interference. c. In situ Measurement: Acquire NMR spectra while applying controlled potentials (constant voltage or constant current) to the carbon electrodes. d. Quantification: Integrate NMR signals to determine the number of ions in the adsorbed (pore) state versus the bulk state as a function of the applied potential. e. Spectral Analysis: Analyze chemical shifts to infer changes in the ion solvation environment and confinement effects [10].
Electrochemical Characterization for CDI & Supercapacitors
- Objective: To determine salt adsorption capacity (SAC), charge efficiency (Λ), and energy consumption.
- Detailed Protocol: a. Cell Assembly: Use a flow-by or flow-through cell architecture with a known mass of porous carbon electrodes, separated by a spacer [16]. b. Operation Modes: * Constant Voltage (CV): Apply a fixed voltage (typically 0.8-1.4 V) for a set time (adsorption), then short-circuit or reverse the voltage for regeneration. Monitor effluent concentration with a conductivity flow cell. * Constant Current (CC): Apply a fixed current. This offers better control over effluent concentration, especially in MCDI configurations [16]. c. Data Analysis: * SAC: Calculate from the integral of the concentration drop over time during adsorption. * Charge Efficiency (Λ): Λ = (F × Γsalt) / Γcharge, where F is Faraday's constant, Γsalt is the salt adsorption capacity, and Γcharge is the total charge transferred. A higher Λ indicates a more dominant counter-ion adsorption mechanism [16].
Computational Approaches
Density Functional Theory (DFT) with Implicit Solvation
- Objective: To calculate adsorption energies of ions in different desolvation states and on doped carbon surfaces.
- Detailed Workflow: a. Model Construction: Build a slab or cluster model of the carbon electrode, which may include topological defects (5-/7-membered rings) or heteroatom dopants (N, O) [13] [18]. b. Solvation Model: Use an implicit solvation model (e.g., VASPsol, ENVIRON) to account for the bulk electrolyte's screening effect [19]. c. Adsorption Energy Calculation: Compute the energy, Eads = E(ion@surface) - Esurface - Eion, where Eion is the energy of the ion in its reference state (e.g., in solution). A more negative Eads indicates stronger, more favorable adsorption [18]. d. Electronic Structure Analysis: Examine charge density differences and Bader charges to understand the nature of the adsorption interaction.
Fully Grand-Canonical (FGC) DFT Approaches
- Objective: To move beyond the computational hydrogen electrode (CHE) model and simulate the electrified interface at a constant electrode potential.
- Detailed Workflow: a. Charged Slab: Perform DFT calculations on a slab model with a non-zero net charge. b. Counter-Charge Model: Introduce a counter-charge in the vacuum/dielectric region (e.g., via an implicit solvation model) to maintain overall cell neutrality [19]. c. Potential Control: Vary the number of electrons in the slab to map the system's energy as a function of electrode potential. d. Analysis: This approach can capture non-Nernstian behavior and non-integer electrosorption valencies, providing a more realistic picture of the potential-dependent electrosorption process [19].
Visualizing Electrosorption Mechanisms and Workflows
The following diagrams illustrate the core concepts and experimental pathways for studying electrosorption mechanisms.
Charge Balancing Mechanisms in Porous Carbon
Experimental Workflow for Mechanism Analysis
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Reagents and Materials for Electrosorption Research
| Item / Solution | Function / Rationale | Example Use Case |
|---|---|---|
| Porous Carbon Electrodes | The core material for electrosorption; its pore size distribution and surface chemistry dictate ionophilicity. | Activated carbon, carbide-derived carbon, carbon aerogels for CDI and supercapacitors [10] [16]. |
| Ion Exchange Membranes | Selectively allow counter-ions to pass while blocking co-ions, enhancing charge efficiency. | Used in MCDI cells to force a counter-ion adsorption mechanism [16]. |
| Aqueous Electrolytes (e.g., Na₂SO₄, KCl) | Provide the ions for electrosorption; their concentration and ion solvation energy are key variables. | Standard electrolytes for studying fundamental mechanisms in water [10] [15]. |
| NMR-Active Isotopes (e.g., ²³Na, ³⁵Cl) | Enable direct tracking of ion populations and environments within pores via in situ NMR. | Quantifying sodium ion adsorption in microporous carbons [10]. |
| Heteroatom Dopant Precursors (e.g., Melamine) | Introduce nitrogen or other heteroatoms into the carbon lattice to modify surface polarity and ion adsorption energy. | Creating N/O-codoped dense porous carbons for enhanced K⁺ adsorption [18]. |
| Implicit Solvation Models (e.g., VASPsol) | Computational tools that approximate the solvent's effect, enabling efficient DFT calculations of solvated ions at interfaces. | Calculating electrosorption valencies and potential-dependent adsorption energies [19]. |
The dichotomy between counter-ion adsorption and co-ion ejection represents a fundamental aspect of charge storage and salt removal in porous carbon electrodes. The dominant mechanism is not predetermined but is an emergent property of the complex interplay between pore size, ion solvation energy, and electrode surface chemistry. The move towards "entropy-driven" or "high-entropy" carbons with designed disorder and multiple dopants further underscores the need to master these fundamentals [13]. A deep understanding of these mechanisms, enabled by the experimental and computational toolkit outlined herein, is essential for rationally designing next-generation materials for energy storage and water purification technologies. By framing system design around the goal of controlling ionophilicity, researchers can push the performance boundaries of supercapacitors and CDI systems.
The Impact of Pore Geometry and Confinement on Ion Hydration and Distribution
The pursuit of high-performance supercapacitors has positioned porous carbon materials as premier electrode candidates, owing to their affordability, high specific surface area, and tunable porosity [20]. The energy storage mechanism in these Electric Double Layer Capacitors (EDLCs) hinges fundamentally on ion adsorption at the electrode-electrolyte interface. This process is profoundly influenced by the nanoscale environment within the electrode's pores. When the pore size approaches the dimensions of the electrolyte ions, confinement effects emerge that drastically alter ion hydration, distribution, and dynamics compared to the bulk phase. Understanding these effects is paramount for advancing ion adsorption research and engineering next-generation porous carbon electrodes with enhanced energy and power densities [21] [22]. This whitepaper synthesizes recent insights into how pore geometry and confinement dictate ion behavior, framing them within the broader context of optimizing ion adsorption in porous carbon electrodes.
Experimental and Simulation Findings on Ion Behavior under Confinement
Structural and Dynamic Alterations in Slit Micropores
Molecular dynamics (MD) simulations of ionic liquid (IL)-organic solvent mixtures confined in slit-like carbon micropores reveal behaviors starkly different from the bulk phase. Studies on mixtures of [EMIM][NTf2] and Dimethyl Sulfoxide (DMSO) demonstrate that the electrolyte's composition within the pore deviates from that in the reservoir, with certain pore sizes exhibiting a preferential increase in counterion concentration upon DMSO dilution [21].
A key finding is the oscillatory dependence of disjoining pressure and pore structure on pore size. As the width of a slit pore increases from 0.7 nm to 1.9 nm, the disjoining pressure oscillates due to abrupt, layered structural rearrangements of the confined electrolyte [21]. The system forms a single ion layer in pores up to 0.9 nm wide, with an additional cation-anion layer inserting with every ~0.4 nm increase in width. This structural quantization leads to correlated oscillations in excess charge at the pore center and the number of hydrogen bonds [21]. Furthermore, the confinement impacts the molecular conformation of ions, altering the distribution of cis and trans conformers of the [NTf2]⁻ anion based on the pore size [21].
The dynamics of ions and solvent molecules are also significantly hampered under confinement. The dynamics of ions and solvents show abrupt changes with different pore sizes, with ion diffusivities in micropores observed to be nearly two orders of magnitude slower than in bulk solutions [21] [20].
Mesoscale Ion Dynamics and Electrosorption
At the mesoscale, the spatiotemporal distribution of electrosorbed ions across a network of nanopores plays a critical role in charge storage, particularly during fast charging. Research using multilayered reduced graphene oxide membranes as model electrodes shows that the electrode's mesostructure—including nanoslit size distribution, pore size distribution, and overall electrode thickness—dynamically influences how ion electrosorption proceeds [22].
During charging, electrosorbed ions mediate both migration and diffusion currents. The response of these currents to different charging rates is governed by the nanoporous electrode's mesostructure. This understanding is vital for designing electrodes that do not sacrifice power density for energy density [22].
Enhancing Performance through Heteroatom Doping and Entropy-Driven Design
Material design strategies directly manipulate the pore geometry and chemical environment to improve ion adsorption. Synthesizing heteroatom-doped porous carbons from ionic liquid precursors is a promising approach. For instance, N, S-doped carbon (SPC-900) derived from ILs achieves a high specific surface area (995.5 m² g⁻¹) and a specific capacitance of 234.8 F g⁻¹ [20]. Density Functional Theory (DFT) research reveals that the synergy between heteroatom doping and C-S-C active sites effectively lowers the adsorption energy of electrolyte ions, thereby strengthening energy storage performance [20].
A novel conceptual framework proposes designing disordered porous carbons from an entropy-driven perspective, leading to "high-entropy carbon materials." This strategy is based on increasing the system's entropy through:
- Unit Entropy: Decreasing the graphene domain size and increasing the number of basic structural units.
- Ring Entropy: Introducing topological defects by distorting the ideal six-membered carbon rings into asymmetric five- or seven-membered rings.
- Element Entropy: Doping multiple non-metallic or metallic elements into the carbon lattice [13]. This entropy-driven approach aims to create a variety of adsorption sites, potentially leading to novel capacitance storage mechanisms [13].
Table 1: Summary of Key Quantitative Findings from Recent Studies on Ion Confinement.
| Study Focus | Material/System | Key Quantitative Finding | Impact on Performance | ||
|---|---|---|---|---|---|
| Layered Structure & Dynamics [21] | [EMIM][NTf2]/DMSO in slit carbon pores | Layered structure changes every ~0.4 nm; dynamics slow by ~2 orders of magnitude. | Determines capacitance oscillation and charging speed. | ||
| Heteroatom Doping [20] | N, S-doped porous carbon (SPC-900) | Specific capacitance of 234.8 F g⁻¹; surface area of 995.5 m² g⁻¹. | Enhances energy density via reduced ion adsorption energy. | ||
| Capacitance Performance [20] | SPC-900 | SPC-900 symmetric cell | Energy density: 6.02 Wh kg⁻¹; Power density: 244.6 W kg⁻¹; 84.15% capacitance retention after 8000 cycles. | Highlights overall device performance and cycle life. |
Detailed Experimental Protocols
Molecular Dynamics (MD) Simulations for Confinement Analysis
Objective: To investigate the structural and dynamic properties of ionic liquid-organic solvent mixtures under confinement in carbon micropores [21].
Methodology:
- System Setup: Model a negatively charged slit-like graphite micropore. The pore width is varied systematically from 0.7 nm to 1.9 nm. The electrolyte is composed of [EMIM][NTf2] ionic liquid dissolved in DMSO at multiple concentrations (e.g., 1 M, 1.5 M, 2 M, 2.5 M).
- Force Field and Parameters: Use a force field for the IL with a charge scaling factor (e.g., 0.8) to account for electronic polarization and enhance simulation accuracy. Employ the OPLS-AA force field for DMSO. Model carbon atoms as Lennard-Jones particles with parameters derived from the literature.
- Simulation Execution: Perform all simulations using a package like GROMACS-2022.2. Run simulations in the NVT ensemble (constant Number of particles, Volume, and Temperature) after an initial energy minimization and equilibration in the NPT ensemble (constant Number of particles, Pressure, and Temperature). Maintain a constant temperature of 298 K using a Nosé-Hoover thermostat.
- Data Analysis:
- Density Profiles: Calculate number and charge density distributions along the pore axis to reveal ion layering.
- Disjoining Pressure: Determine the pressure exerted by the confined fluid perpendicular to the pore walls.
- Dynamics: Compute mean-squared displacement (MSD) and self-diffusion coefficients for ions and solvent molecules.
- Structural Analysis: Analyze hydrogen bonding, ion coordination, and molecular conformations (e.g., cis/trans for [NTf2]⁻).
Synthesis of Heteroatom-Doped Porous Carbon
Objective: To fabricate heteroatom-adaptive hierarchical porous carbon materials from ionic liquid precursors via a one-step pyrolysis method [20].
Methodology:
- Precursor Preparation: Synthesize the ionic liquid precursor, for example, by combining dimethylimidazole with acids like sulfuric acid (H₂SO₄) or phosphoric acid (H₃PO₄) to form ILs with [HSO₄]⁻ or [H₂PO₄]⁻ anions, respectively.
- Pyrolysis (Calcination): Place the ionic liquid precursor in a tubular furnace. Pyrolyze under an inert atmosphere (e.g., nitrogen or argon gas). Employ a controlled heating ramp (e.g., 5 °C min⁻¹) to a target temperature (e.g., 900 °C) and maintain this temperature for a set duration (e.g., 2 hours) to achieve carbonization and pore formation. The ionic liquid acts as the carbon source, heteroatom dopant, and self-template pore-forming agent.
- Post-processing: After the furnace cools to room temperature, collect the resulting black carbon product. Wash the product with ethanol and deionized water to remove impurities. Dry the final heteroatom-doped porous carbon (e.g., SPC-900 for sulfate-derived carbon at 900°C) in an oven overnight.
- Characterization:
- Morphology: Use Scanning Electron Microscopy (SEM) and Transmission Electron Microscopy (TEM) to analyze the porous architecture.
- Surface Area and Porosity: Determine the specific surface area and pore size distribution using N₂ adsorption/desorption isotherms and the Brunauer-Emmett-Teller (BET) method.
- Chemical Composition: Identify the types and bonding states of heteroatoms (N, S, P) using X-ray Photoelectron Spectroscopy (XPS).
- Electrochemical Performance: Evaluate the capacitive performance in a three-electrode cell and a symmetric two-electrode cell using cyclic voltammetry, galvanostatic charge-discharge, and electrochemical impedance spectroscopy.
Table 2: Key Research Reagents and Materials for Ion Adsorption Studies.
| Reagent/Material | Function in Research | Technical Context |
|---|---|---|
| Ionic Liquids (e.g., [EMIM][NTf2]) | Primary electrolyte or precursor for doped carbon. | Provides ions for the double layer; serves as a source of heteroatoms (N, S) in carbon synthesis [21] [20]. |
| Aprotic Solvents (e.g., DMSO, ACN) | Diluent for ionic liquid electrolytes. | Reduces viscosity, improves ionic conductivity, and modulates ion-ion interactions [21]. |
| Heteroatom-Doped Porous Carbons | Model electrode material. | Increases conductivity, introduces pseudocapacitance, and lowers ion adsorption energy [13] [20]. |
| Nanoporous Electrodes (e.g., rGO membranes) | Model system for studying mesoscale dynamics. | Allows monitoring of spatiotemporal ion distribution and potential during fast charging [22]. |
Visualizing Core Concepts and Workflows
The following diagrams illustrate the key relationships and experimental workflows discussed in this whitepaper.
Synthesis, Functionalization, and Application of Advanced Porous Carbons
The escalating demand for high-performance energy storage systems has driven extensive research into advanced electrode materials. Porous carbon electrodes, particularly those derived from biomass, have emerged as a cornerstone technology for supercapacitors and batteries, where energy storage occurs primarily through ion adsorption at the electrode-electrolyte interface [23]. The performance of these carbon materials is intrinsically linked to their textural properties and surface chemistry, which govern ion transport, adsorption kinetics, and overall capacitance [23]. Biomass-derived porous carbons represent a sustainable alternative to traditional materials, offering advantages such as low cost, abundance, renewability, and reduced environmental impact [24] [25]. Furthermore, the strategic incorporation of nitrogen heteroatoms into the carbon matrix has proven highly effective for enhancing electrochemical performance through improved surface polarity, electrical conductivity, and additional pseudocapacitive contributions [25] [26]. This technical guide examines current synthesis methodologies, characterization techniques, and performance outcomes for biomass-derived nitrogen-doped porous carbons, contextualized within the fundamental principles of ion adsorption in porous electrodes.
Sustainable Synthesis of Biomass-Derived Porous Carbons
Biomass Precursor Selection and Composition
The selection of biomass precursor is critical, as its inherent composition directly influences the structural properties and performance of the resulting carbon material. Lignocellulosic biomass primarily consists of cellulose, hemicellulose, and lignin, each contributing differently to the final carbon structure [24]. Cellulose-rich precursors tend to form highly fibrous and porous structures, while lignin's high carbon content and aromatic structure yield thermally stable carbons [24]. The presence of proteins in some biomass can lead to self-doping of nitrogen, enhancing CO₂ capture and electrochemical properties through the formation of pyridinic-N and pyrrolic-N functionalities during pyrolysis [24]. Precursors with low ash content, such as wood and nut shells, are generally preferred for obtaining carbon materials with well-developed porosity [24].
Numerous biomass sources have been successfully utilized, including rotten wood [27], reed straw [26], tobacco straw [28], daikon [29], and various agricultural wastes. These precursors provide a renewable and often low-cost feedstock for carbon production, contributing to a circular economy approach while reducing reliance on fossil-based resources.
Synthesis and Activation Methods
The conversion of biomass into porous carbon typically involves thermal treatment under inert atmosphere, often followed by activation to develop porosity and increase specific surface area.
Table 1: Common Synthesis Methods for Biomass-Derived Porous Carbons
| Method | Process Conditions | Key Features | Resulting Material Properties |
|---|---|---|---|
| Pyrolysis | Thermal decomposition in inert atmosphere (400-900°C) | Base carbonization process; determines fundamental carbon structure | Char/biochar with basic carbon framework |
| Hydrothermal Carbonization (HTC) | Aqueous suspension at elevated temperatures (180-250°C) and autogenous pressure | Converts biomass into hydrochar; preserves functional groups | Carbon spheres with oxygen-containing surface groups |
| Chemical Activation | Impregnation with activators (KOH, NaOH, H₃PO₄, ZnCl₂) followed by pyrolysis | Creates microporosity; significantly increases surface area | High specific surface area (up to 3000 m²/g); microporous dominance |
| Physical Activation | Treatment with oxidizing gases (CO₂, H₂O steam) at high temperatures | Develops porosity through gasification; less corrosive than chemical methods | Broader pore size distribution; less microporous than chemical activation |
Chemical activation is particularly effective for creating microporous structures with high specific surface areas. For instance, KOH activation creates micropores through redox reactions and potassium intercalation, effectively etching the carbon framework [24]. The activation temperature significantly influences pore development, with higher temperatures (700-900°C) generally promoting increased surface area and pore volume [26].
An innovative approach involves leveraging natural fungal decay in rotten wood, which creates intrinsic porous structures through microbial activity. This method can be combined with chemical activation to produce hierarchical porous carbons with specific surface areas exceeding 1200 m²/g [27].
Nitrogen Doping Strategies and Mechanisms
Nitrogen doping has emerged as a powerful strategy to enhance the electrochemical performance of porous carbons by modifying their electronic structure, surface chemistry, and catalytic activity. The incorporation of nitrogen creates favorable sites for ion adsorption and facilitates Faradaic reactions, thereby increasing overall capacitance [25] [26].
Nitrogen Doping Methods
Table 2: Nitrogen Doping Strategies for Biomass-Derived Porous Carbons
| Method | Process Description | Advantages | Nitrogen Content Achieved |
|---|---|---|---|
| In-Situ Doping | Direct pyrolysis of N-rich biomass (e.g., chitosan, algae, glucosamine) or addition of N-precursors (melamine, urea) before carbonization | Uniform nitrogen distribution; simple one-step process | Varies with precursor (typically 2-6%) |
| Post-Synthesis Doping | Treatment of pre-carbonized biomass with ammonia or nitrogen plasma at elevated temperatures | Precise control over nitrogen functionality; higher nitrogen incorporation | Can exceed 10% with optimized conditions |
| Combined Activation-Doping | Simultaneous chemical activation and nitrogen doping using agents like NH₃ or mixtures (e.g., KOH + melamine) | Integrated process; creates porous N-doped structures in single step | Moderate to high (3-8%) depending on conditions |
In-situ doping during pyrolysis is particularly advantageous for biomass-derived carbons. For example, mixing reed straw with melamine prior to pyrolysis yielded N-doped carbon with 6.02% nitrogen content and a specific surface area of 547.1 m²/g [26]. Similarly, treatment of daikon-derived carbon with ammonia at 900°C created a material with excellent electrocatalytic performance for oxygen reduction reaction [29].
Nitrogen Functionality and Its Effects
The electrochemical benefits of nitrogen doping depend not only on the total nitrogen content but also on the specific bonding configurations within the carbon matrix:
- Pyridinic-N: Contributes to pseudocapacitance through Faradaic reactions and provides active sites for metal ion adsorption; particularly effective for enhancing specific capacitance [26].
- Pyrrolic-N: Similar to pyridinic-N, participates in redox reactions and improves electrochemical performance.
- Graphitic-N: Enhances electronic conductivity through donation of electrons to the carbon π-system, facilitating charge transfer [25].
- Pyridinic-N-Oxide: May influence surface polarity and wettability.
Theoretical calculations indicate that pyrrolic-N and pyridinic-N exhibit stronger binding energies to Li⁺ ions (4.46 eV and 4.26 eV, respectively) compared to pristine graphite (3.64 eV), significantly enhancing ion adsorption capacity [28].
Structural Characterization and Property Analysis
Comprehensive characterization of biomass-derived porous carbons is essential for correlating material properties with electrochemical performance.
Textural Properties and Pore Structure
Nitrogen adsorption-desorption analysis provides critical information about specific surface area, pore volume, and pore size distribution. Biomass-derived carbons typically exhibit Type I isotherms characteristic of microporous materials, often with H4-type hysteresis loops indicating the presence of mesoporosity [28]. The pore architecture significantly influences ion transport and accessibility, with hierarchical structures containing micro-, meso-, and macropores demonstrating superior electrochemical performance [29].
Table 3: Textural Properties of Selected Biomass-Derived Porous Carbons
| Biomass Precursor | Synthesis Method | Specific Surface Area (m²/g) | Pore Characteristics | Reference |
|---|---|---|---|---|
| Rotten Wood | KOH activation + EDA nitrogen source | 1204 | Hierarchical porous structure | [27] |
| Reed Straw | Pyrolysis with melamine (1:3 ratio) + KOH activation | 547.1 | Micro- and mesoporous structure | [26] |
| Tobacco Straw | NaOH activation + melamine doping | 378.5 | Predominantly micropores | [28] |
| Daikon | NH₃ activation at 900°C | Not specified | Hierarchical porous architecture | [29] |
| Wood | Combined chemical and physical activation | Pore diameter enlarged from 0.54 nm to 1.13 nm | Tailored pore channels | [17] |
Chemical and Structural Characterization
X-ray photoelectron spectroscopy (XPS) reveals the chemical composition and nitrogen bonding configurations in doped carbons. For instance, XPS analysis of reed straw-derived carbon confirmed the presence of pyridinic-N, pyrrolic-N, and graphitic-N species, with pyridinic-N identified as particularly active for charge storage [26].
Raman spectroscopy typically shows characteristic D and G bands around 1350 cm⁻¹ and 1580 cm⁻¹, respectively, with the intensity ratio (ID/IG) providing information about defect density in the carbon structure. Nitrogen doping generally increases defect concentration, which can enhance electrochemical activity [28].
X-ray diffraction (XRD) patterns of biomass-derived carbons often display broad peaks around 24° and 43°, corresponding to the (002) and (100) planes of graphitic carbon, indicating predominantly amorphous or turbostratic structures with limited crystallinity [26].
Electrochemical Performance in Energy Storage Devices
Supercapacitor Applications
Biomass-derived N-doped porous carbons have demonstrated exceptional performance as electrode materials for supercapacitors, particularly in electric double-layer capacitors (EDLCs) where charge storage occurs through ion adsorption at the electrode-electrolyte interface [23].
Table 4: Electrochemical Performance of Biomass-Derived N-Doped Porous Carbons in Supercapacitors
| Biomass Precursor | Specific Capacitance (F/g) | Test Conditions | Cycling Stability | Reference |
|---|---|---|---|---|
| Rotten Wood | 448 F/g | 0.2 A/g in 3-electrode system | 95% retention after 10,000 cycles | [27] |
| Reed Straw (with melamine) | 202.8 F/g at 1 A/g; 158 F/g at 20 A/g | 1 A/g in 6 M KOH | 96.3% retention after 5,000 cycles at 20 A/g | [26] |
| Wood-derived (for Zn-ion hybrid SC) | 412.76 F/g (3-electrode); 269.54 mAh/g at 0.2 A/g | 5 mV/s in 3-electrode system | 93.55% retention after 20,000 cycles | [17] |
| Coal-derived (optimized with O-groups) | 273 F/g | Aqueous electrolyte | High stability | [15] |
The enhanced capacitance of N-doped carbons arises from both electric double-layer formation and pseudocapacitive contributions from nitrogen functional groups. Pyridinic and pyrrolic nitrogen species undergo reversible redox reactions, providing Faradaic capacitance in addition to the electrostatic charge storage of EDLCs [26]. This combination enables both high power density and improved energy density.
Battery Applications
In lithium-ion batteries, N-doped biomass-derived carbons serve as effective anode materials. Tobacco straw-derived N-doped carbon (TsNC) delivered a remarkable reversible specific capacity of 475.9 mA h g⁻¹ at 60 mA g⁻¹ after 500 cycles, significantly outperforming its undoped counterpart [28]. This enhancement is attributed to the increased lithium-ion adsorption sites provided by nitrogen functional groups, particularly graphitic-N, pyrrolic-N, and pyridinic-N.
Kinetic analysis revealed a predominant surface capacitive-controlled behavior in N-doped carbons, facilitating rapid charging and discharging at high rates [28]. This characteristic is particularly valuable for applications requiring high power density and fast charging capabilities.
Advanced Concepts: Ion Desolvation and Pore Engineering
A fundamental understanding of ion desolvation processes is crucial for optimizing carbon electrodes for specific electrolytes. Recent research has revealed that potassium ions can exist in five distinct desolvation states ([K(H₂O)₀₋₄]⁺) within porous carbon electrodes, each with different desolvation energies and diffusion barriers [15]. This insight provides a theoretical foundation for designing pore structures that minimize ion transport resistance and maximize capacitance.
Pore size optimization is particularly critical when dealing with hydrated ions. For instance, in zinc-ion hybrid supercapacitors, there is often a size discrepancy between carbon cathode pores and the [Zn·(H₂O)₆]²⁺ complex (diameter ∼0.86 nm) [17]. Strategic pore engineering through combined chemical and physical activation can successfully enlarge pore diameters to better accommodate hydrated ions, significantly enhancing ionic migration kinetics and energy density [17].
The relationship between pore size and capacitance follows a non-monotonic trend. When pore sizes approach 1 nm, normalized capacitance decreases, but drops sharply when pores become smaller than the solvated ion size due to distortion of solvation shells, allowing closer ion approach to the electrode surface [23]. Molecular dynamics simulations suggest that capacitance strongly correlates with charge compensation per carbon rather than geometric properties alone, highlighting the importance of electronic structure in addition to porosity [23].
Experimental Protocols
Representative Synthesis Procedure: N-Doped Carbon from Reed Straw
Materials: Reed straw, melamine, KOH, HCl (for washing), deionized water.
Equipment: Tube furnace, quartz boat, grinding apparatus, drying oven.
Procedure:
- Pre-treatment: Wash reed straw thoroughly with deionized water and dry at 100°C for 12 hours. Grind into fine powder.
- Mixing: Mix reed straw powder with melamine at mass ratio of 3:1 (reed straw:melamine). Add KOH as chemical activator (typical ratio: 1:1-3, carbon:activator).
- Pyrolysis: Transfer mixture to quartz boat and place in tube furnace. Heat under N₂ atmosphere (flow rate: 200 mL/min) to 550°C with heating rate of 5°C/min. Hold at target temperature for 1-2 hours.
- Post-treatment: After cooling to room temperature, wash product repeatedly with diluted HCl to remove impurities and neutralize pH. Rinse with deionized water until neutral pH is achieved.
- Drying: Dry final product at 100°C for 12 hours to obtain N-doped porous carbon [26].
Representative Synthesis Procedure: N-Doped Carbon from Tobacco Straw
Materials: Tobacco straw, melamine, NaOH, HCl, deionized water.
Equipment: Tube furnace, agate mortar, drying oven, washing apparatus.
Procedure:
- Pre-carbonization: Cut tobacco straw into pieces, wash, and dry at 100°C for 12 hours. Grind into powder. Heat to 500°C under N₂ atmosphere for 2 hours (heating rate: 5°C/min).
- Mixing: Mix pre-carbonized powder with NaOH and melamine in 1:1:1 mass ratio. Grind in agate mortar for 5 minutes to ensure homogeneity.
- Activation/Doping: Heat mixture in tube furnace under N₂ atmosphere to 600°C for 1 hour (heating rate: 5°C/min).
- Post-treatment: Wash resulting product multiple times with diluted HCl and deionized water to remove impurities and achieve neutral pH.
- Drying: Dry final product at 100°C for 12 hours [28].
Research Reagent Solutions
Table 5: Essential Research Reagents for Biomass-Derived Porous Carbon Synthesis
| Reagent | Function | Application Examples |
|---|---|---|
| KOH | Chemical activator; creates microporosity through etching | Universal activation agent for high surface area carbons [27] [26] |
| Melamine | Nitrogen source for in-situ doping | Reed straw and tobacco straw nitrogen doping [26] [28] |
| Ammonia (NH₃) | Nitrogen source for post-synthesis doping | Gas-phase doping of daikon-derived carbon [29] |
| NaOH | Chemical activator; alternative to KOH | Tobacco straw activation [28] |
| Ethylenediamine (EDA) | Nitrogen source and activator | Enhancing nitrogen content in rotten wood-derived carbon [27] |
| H₃PO₄ | Chemical activator; creates mesoporosity | Softer activation compared to alkalis [24] |
Biomass-derived N-doped porous carbons represent a promising class of sustainable materials for advanced energy storage applications. Their performance in ion adsorption and electrochemical energy storage is governed by an intricate interplay between textural properties (specific surface area, pore size distribution, pore volume) and surface chemistry (heteroatom doping, functional groups). The strategic incorporation of nitrogen functionalities, particularly pyridinic and pyrrolic nitrogen, significantly enhances capacitance through combined electric double-layer and pseudocapacitive charge storage mechanisms.
Future research directions should focus on precise pore engineering to match specific hydrated ion sizes, advanced doping strategies to control nitrogen functionality distribution, and scalable synthesis methods to facilitate commercial implementation. The integration of theoretical modeling with experimental approaches will further advance our understanding of ion adsorption/desolvation processes in confined porous structures, enabling the rational design of next-generation carbon electrodes with enhanced performance characteristics.
Diagrams
Diagram 1: Synthesis workflow for biomass-derived N-doped porous carbon, showing key steps and structural outcomes.
Diagram 2: Mechanisms of performance enhancement through nitrogen doping in porous carbon electrodes.
The performance of electrochemical and adsorption systems—spanning energy storage devices like supercapacitors and hybrid capacitors to environmental remediation technologies for heavy metal removal—is fundamentally governed by the interaction between specific ions and the porous carbon electrodes they encounter. The central challenge in this field lies in the frequent mismatch between the dimensions of pore channels within carbon materials and the size of the target ions, whether solvated or desolvated. This discrepancy can severely compromise ionic migration kinetics, leading to diminished energy density, limited storage capacity, and reduced adsorption efficiency [17] [30]. A critical finding from recent research highlights that for efficient storage of zinc ions (e.g., [Zn·(H2O)6]2+ with a diameter of ~0.86 nm), carbon cathode pores must be appropriately sized to facilitate ion access and adsorption [17].
Hierarchical pore engineering has emerged as a powerful strategy to overcome these limitations. This approach involves the deliberate design of carbon materials that incorporate a synergistic blend of micropores (for high ion storage capacity), mesopores (for efficient ion transport), and macropores (for rapid ion buffering). The ultimate goal is to tailor both the porosity and surface chemistry of carbon materials to achieve optimal, ion-specific performance. This technical guide delves into the advanced methods of activators and pyrolysis used to precisely tune these hierarchical pore structures, providing a comprehensive resource for researchers and scientists working at the intersection of materials science, electrochemistry, and environmental technology.
The Science of Ion Adsorption in Porous Carbons
Fundamental Mechanisms
Ion adsorption at solid–water interfaces is a complex process crucial for the operation of supercapacitors, water desalination, and electrocatalysis. The classic Electric Double Layer (EDL) model is often insufficient to fully describe the behavior in complex, disordered porous carbons. Modern studies emphasize the Modified Donnan Model, which more accurately accounts for the distinct ways charges are adsorbed within the volume of micro-, meso-, and macropores [31] [30].
Crucially, the adsorption process is not a simple, single mechanism. Upon charging, three primary phenomena occur almost simultaneously: co-ion desorption (expulsion of ions with the same charge as the electrode), counter-ion adsorption (uptake of ions with the opposite charge), and ion exchange (a mutual exchange of co- and counter-ions) [31]. The kinetics and equilibrium of these processes are heavily influenced by the pore structure and the surface chemistry of the carbon material.
The role of water itself, particularly in aqueous electrolytes, cannot be overlooked. The hydronium (H3O+) and hydroxide (OH–) ions can compete with electrolyte salt ions to act as charge-compensating species. For instance, under acidic conditions, NMR spectroscopy has revealed that more TFSI– anions may be adsorbed in carbon pores than Li+ cations, with the charge imbalance being compensated by the specific adsorption of H3O+ ions [30]. This highlights that the electrolyte pH and the surface charge of the carbon (its Point of Zero Charge, PZC) are critical parameters that directly influence ion uptake and the overall capacitance of the system.
The Critical Role of Ion-Pore Size Matching
A quintessential example of the pore-ion size mismatch problem is found in Zinc-Ion Hybrid Supercapacitors (ZiHSCs). The common charge carrier, the hydrated [Zn·(H2O)6]2+ complex, has a diameter of approximately 0.86 nm [17]. If the dominant pores in the cathode carbon are significantly smaller than this diameter, the ionic migration kinetics are drastically weakened, leading to low energy density.
Research has demonstrated that successfully enlarging the pore diameters of a wood-derived carbon from 0.54 nm to 1.13 nm directly addressed this mismatch. This structural optimization resulted in a remarkable specific capacity of 269.54 mAh/g and an energy density of 210.76 Wh/kg, coupled with exceptional cycling stability (93.55% capacity retention after 20,000 cycles) [17]. This case underscores that pore size is not merely a contributor but a decisive factor in achieving high performance.
Table 1: Target Ion Sizes and Corresponding Optimal Pore Dimensions
| Ion Species | Ion Diameter (nm) | Recommended Pore Size (nm) | Key Application |
|---|---|---|---|
Hydrated Zinc Ion [Zn·(H2O)6]2+ |
~0.86 [17] | >0.86 (e.g., 1.13 nm) [17] | Zinc-Ion Hybrid Capacitors |
| Typical Aqueous Electrolyte Ions | Varies (solvated/desolvated) | Hierarchical: Micropores (<2 nm) for storage, Mesopores (2-50 nm) for transport [32] | Electric Double-Layer Capacitors |
| Cadmium Ions (Cd²⁺) | N/A | Smaller crystallite size & high surface area favored (e.g., 51.66 nm crystallites) [33] | Heavy Metal Adsorption |
Activation Methods for Hierarchical Pore Engineering
The creation of a hierarchical pore structure is predominantly achieved through activation processes, which can be broadly classified into physical and chemical methods. The choice of activator and pyrolysis conditions directly dictates the final pore characteristics.
Chemical Activation
Chemical activation is a single-step process that involves impregnating a carbon precursor with a chemical agent followed by pyrolysis under an inert atmosphere. The chemical agent acts as a dehydrating and oxidizing agent, inhibiting tar formation and promoting the development of porosity.
Alkaline Hydroxides (KOH and NaOH): These are among the most effective activators for generating ultra-high surface areas. The activation mechanism involves redox reactions and potassium/sodium intercalation into the carbon lattice, which violently etches the carbon to create micropores and mesopores.
- KOH Activation: A classic, powerful method. Its etching reaction is
6KOH + 2C → 2K + 3H2 + 2K2CO3. The subsequent decomposition of K2CO3 further releases CO2, contributing to additional porosity. - NaOH Activation: Emerging as a more sustainable and cost-effective alternative to KOH. When applied to hemp hurd biochar at a 1:4 ratio (biochar:NaOH), it produced a specific surface area (SSA) of 3033 m²/g and a mesopore-to-micropore volume ratio of 1.58 [34]. This balanced structure yielded a high specific capacitance of 725 F/g in a three-electrode system with 1 M H2SO4 electrolyte [34].
- KOH Activation: A classic, powerful method. Its etching reaction is
Carbonate Salts (K₂CO₃ and ZnCO₃): These are considered milder and less corrosive activators compared to hydroxides, but they are highly effective for creating hierarchical structures.
- K₂CO₃ Activation: The decomposition
K2CO3 → K2O + CO2and the subsequent reduction of K2O by carbon (K2O + C → 2K + CO) generate metallic potassium, which expands the carbon lattice and creates pores. The released CO2 also acts as a physical oxidant. - Bicarbonate/Carbonate Dual Activation: A innovative strategy using K₂CO₃ and ZnCO₃ on tobacco stems and lignin produced carbon with an SSA of 2154 m²/g, a mesopore proportion of 27.6%, and a high surface oxygen content of 14.04 at.% [35]. The ZnCO₃ served a dual role as both an activator and a template, with its decomposition product (ZnO) undergoing carbothermal reduction to create additional mesopores. This material delivered a specific capacitance of 376 F/g as a cathode in zinc-ion hybrid capacitors [35].
- K₂CO₃ Activation: The decomposition
Table 2: Comparison of Common Chemical Activators for Hierarchical Pores
| Activator | Typical Pyrolysis Temperature | Key Pore Structure Outcomes | Advantages & Disadvantages |
|---|---|---|---|
| KOH | 600-800 °C | Ultra-high SSA (>3000 m²/g), predominantly microporous [34] | Pro: Exceptible SSA. Con: Highly corrosive, expensive. |
| NaOH | ~720 °C [34] | High SSA (>3000 m²/g), tunable V~meso~/V~micro~ ratio [34] | Pro: Less corrosive & costly than KOH. Con: Slightly lower yields. |
| K₂CO₃ | 800-900 °C | Develops micro- and mesopores, good hierarchical structure [35] [36] | Pro: Milder, less corrosive. Con: May require higher temperatures. |
| ZnCO₃ / ZnCl₂ | ~800 °C [35] | Effective for creating mesopores, acts as a template [35] | Pro: Good mesoporosity, templating effect. Con: Can leave residues. |
Physical Activation and Combined Approaches
Physical activation involves the gasification of a pre-carbonized "char" using oxidizing gases such as steam (H₂O) or carbon dioxide (CO₂) at high temperatures (800-1100 °C). The reaction C + H2O → H2 + CO etches the carbon surface, primarily creating micropores. A key advantage is the ability to use the marginal carbon atoms in pre-existing pores to selectively enlarge them. For instance, a combined chemical and physical (H₂O steam) activation process was shown to successfully enlarge pore diameters from 0.54 nm to 0.71 nm and 1.13 nm, which was pivotal for accommodating zinc ions [17].
Combined physicochemical activation leverages the strengths of both methods. An initial chemical activation can create a foundational porous network, which a subsequent physical activation step can further refine and widen, offering unparalleled control over the final pore size distribution.
Experimental Protocols: A Detailed Workflow
This section provides a detailed, actionable methodology for synthesizing hierarchical porous carbons via chemical activation, based on protocols refined in recent literature.
Objective: To synthesize high-surface-area porous carbon with a balanced micro/mesopore structure for supercapacitor electrodes.
Materials:
- Precursor: Hemp hurd (or other lignin-rich biomass).
- Activator: Sodium Hydroxide (NaOH), analytical grade.
- Gases: High-purity Nitrogen (N₂) and Argon (Ar) for inert atmospheres.
- Other Chemicals: 1 M Hydrochloric Acid (HCl) for washing, deionized water.
Procedure:
- Precursor Preparation: Dry raw hemp hurd in an oven overnight to remove moisture. Chop into small pieces (approx. 5-20 mm).
- Pyrolysis Carbonization:
- Load ~20 g of dried hemp hurd into a tubular furnace.
- Heat to 500 °C with a ramping rate of 5 °C/min under a continuous N₂ flow (500 sccm).
- Hold at 500 °C for 2 hours to produce biochar (Hurd-BC).
- Allow to cool to room temperature under N₂ flow.
- Biochar Processing: Grind the resulting biochar into a fine powder using a ball mill (e.g., 20 min at 30 Hz).
- Chemical Activation:
- Mechanically mix the ground Hurd-BC with solid NaOH powder at the desired weight ratio (e.g., 1:1, 1:2, 1:3, 1:4) in a mortar.
- Transfer the mixture to a reactor and heat in a tubular furnace under an Ar atmosphere.
- Heat to 720 °C at 5 °C/min and maintain for 1 hour.
- Post-Activation Processing:
- Cool the activated product to room temperature.
- Wash the product by stirring in 1 M HCl solution at 100 °C for 4 hours to remove inorganic salts and metal impurities.
- Rinse repeatedly with deionized water until the filtrate reaches a neutral pH.
- Collect the final product by filtration and dry in an oven at 100 °C overnight.
The following workflow diagram visualizes this synthesis and characterization pipeline:
Objective: To fabricate oxygen-rich hierarchical porous carbon from waste biomass for enhanced zinc ion storage.
Materials:
- Carbon Precursors: Tobacco stems (TS) and enzymatically hydrolyzed lignin (EHL).
- Dual Activators: Potassium Carbonate (K₂CO₃) and Zinc Acetate Dihydrate ((CH₃COO)₂Zn·2H₂O).
- Other: Deionized water, HCl.
Procedure:
- Precursor Mixing: Combine tobacco stems and enzymatically hydrolyzed lignin with an aqueous K₂CO₃ solution to form a homogeneous mixture. The solubility of lignin in K₂CO₃ improves compatibility.
- In-situ Co-precipitation: Add a zinc acetate solution to the mixture. The Zn²⁺ ions co-precipitate with CO₃²⁻, converting part of the K₂CO₃ into ZnCO₃ within the carbon matrix.
- Drying: Dry the mixture thoroughly to obtain a precursor composite.
- Pyrolysis & Activation: Heat the precursor to 800 °C under an inert atmosphere. During this step:
- K₂CO₃ decomposes and activates the carbon, primarily creating micropores.
- ZnCO₃ decomposes to ZnO and CO₂. The ZnO is subsequently reduced by carbon (carbothermal reduction:
ZnO + C → Zn + CO), generating metallic zinc vapor which acts as a porogen to create mesopores.
- Washing and Drying: The resulting carbon is washed with HCl to remove residual zinc and inorganic salts, followed by rinsing with water and drying.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Hierarchical Porous Carbon Synthesis
| Reagent / Material | Function / Role in Synthesis | Key Consideration |
|---|---|---|
| KOH / NaOH Pellets | Powerful chemical activator; creates ultra-high surface area and microporosity. | Highly corrosive; requires careful handling. NaOH is a cost-effective alternative [34]. |
| K₂CO₃ / ZnCO₃ | Milder chemical activator; effective for creating hierarchical micro-mesoporous structures [35] [36]. | K₂CO₃ is less corrosive; ZnCO₃ provides a templating effect for mesopores [35]. |
| Lignin-Rich Biomass | Sustainable carbon precursor; high natural carbon content contributes to yield and structure [34] [35]. | Hemp hurd, tobacco stems, enzymatically hydrolyzed lignin are effective precursors [34] [35]. |
| Inert Gas (N₂, Ar) | Creates an oxygen-free atmosphere during pyrolysis/activation, preventing combustion. | High purity (>99.99%) is essential to avoid side reactions and ensure sample integrity. |
| HCl Solution | Post-synthesis washing to remove activation-derived salts, metals, and impurities. | Concentration (e.g., 1M) and washing temperature (e.g., 100°C) impact purification efficiency [34]. |
Characterization and Performance Evaluation
Rigorous characterization is vital to correlate the synthesized material's structure with its performance.
Structural and Chemical Characterization
- Gas Sorption Analysis (BET): Used to determine the Specific Surface Area (SSA), total pore volume, and pore size distribution (PSD). A type I isotherm indicates microporosity, while a type IV isotherm with a hysteresis loop suggests the presence of mesopores [34] [36].
- Scanning Electron Microscopy (SEM) / Transmission Electron Microscopy (TEM): Reveals the surface morphology, internal porous structure (e.g., interconnected multi-cavity designs), and overall architecture of the carbon material [36].
- X-ray Diffraction (XRD): Assesses the degree of crystallinity. Most activated carbons show broad peaks, characteristic of amorphous structures [33] [36].
- Raman Spectroscopy: Quantifies the level of structural defects. The intensity ratio of the D band (defects) to the G band (graphitic carbon), I~D~/I~G~, is a key metric, where a higher ratio indicates more defects, which can be beneficial for ion adsorption [36].
- X-ray Photoelectron Spectroscopy (XPS): Identifies and quantifies surface elemental composition and heteroatom functional groups (e.g., oxygen, nitrogen), which are critical for pseudocapacitance and surface wettability [30] [35].
Electrochemical and Adsorption Performance Metrics
The efficacy of the tuned porous carbon is evaluated based on application-specific metrics.
Table 4: Key Performance Indicators for Different Applications
| Application | Key Performance Metrics | Exemplary Performance from Literature |
|---|---|---|
| Supercapacitors & \nZinc-Ion Hybrid Capacitors | Specific Capacitance (F/g): Charge storage per mass.Energy Density (Wh/kg): Stored energy per mass.Power Density (W/kg): Rate of charge/discharge.Cycle Stability: Capacity retention over cycles. | Capacitance: 725 F/g (Hemp/H₂SO₄) [34].Energy Density: 210.76 Wh/kg (Wood-derived/Zn) [17].Stability: 93.55% after 20,000 cycles [17]. |
| Capacitive Deionization (CDI) | Desalination Capacity (mg/g): Salt adsorption per mass.Adsorption Rate (mg/g/min): Speed of salt removal.Cycle Stability: Retention of capacity over cycles. | Capacity: 114.25 mg/g for NaCl [36].Rate: 6.57 mg/g/min [36].Stability: 95% after 50 cycles [36]. |
| Heavy Metal Adsorption | Uptake Capacity (mg/g): Metal ion adsorbed per mass.Regeneration Efficiency: % recovery after desorption.Cycle Stability: Uptake retention over cycles. | Capacity: 280.11 mg/g for Cd²⁺ [33].Regeneration: ~100% with 3M HCl [33]. |
The following diagram illustrates the complex charge storage mechanisms that occur within a hierarchically porous carbon electrode, combining EDL formation and surface redox reactions:
The strategic use of activators and controlled pyrolysis represents a mature and highly effective methodology for tuning the hierarchical pore structures of carbon materials toward specific ion targets. The evidence is clear: matching pore dimensions to ion size, as demonstrated with zinc ions, and engineering a balanced network of micro-, meso-, and macropores directly translates to superior performance in energy storage and adsorption applications.
Future advancements in this field will likely focus on several key areas:
- Predictive Synthesis: Leveraging machine learning and computational modeling to predict the optimal precursor-activator-pyrolysis combination for a desired pore structure and ion target, moving beyond trial-and-error approaches.
- Atomic-Level Tuning: Greater emphasis on the precise control of surface chemistry (heteroatom doping, functional groups) in conjunction with porosity to exploit synergistic effects for both enhanced EDL formation and pseudocapacitance.
- Advanced In-Situ/Operando Characterization: Using techniques like in-situ XRD and Raman spectroscopy to dynamically observe ion adsorption/desorption and phase transitions within pores during operation, providing unprecedented insight into charge storage mechanisms [17].
- Sustainability: Continued prioritization of waste biomass precursors and the development of "greener," less corrosive activation processes (e.g., using NaOH or carbonates) to reduce the environmental footprint of porous carbon production [34] [35].
By systematically applying the principles and protocols outlined in this guide, researchers can design and fabricate next-generation porous carbon materials with tailored architectures that meet the exacting demands of modern electrochemical and environmental technologies.
In the development of advanced porous carbon electrodes for energy storage systems, understanding the intricate relationship between material properties and electrochemical performance is paramount. The adsorption and desorption of ions within the complex pore networks of carbon electrodes fundamentally govern the efficiency, capacity, and cycling stability of devices such as supercapacitors and ion batteries. This technical guide provides an in-depth examination of four cornerstone characterization techniques—BET surface area analysis, FT-IR spectroscopy, SEM, and elemental analysis—framed within the context of ion adsorption research for porous carbon electrodes. By elucidating the specific capabilities, methodologies, and interpretations of each technique, this document serves as a comprehensive resource for researchers and scientists engaged in the rational design and optimization of next-generation energy storage materials.
BET Surface Area Analysis
Theoretical Principles and Relevance
The Brunauer-Emmett-Teller (BET) theory is the foundational model for determining the specific surface area of solid or porous materials by quantifying the physical adsorption of gas molecules on a solid surface [37]. For porous carbon electrodes involved in ion adsorption, the BET surface area provides critical information as it directly influences the electrode-electrolyte interface area, where energy storage occurs. Materials with engineered pore structures, such as activated carbons, can exhibit exceptionally high surface areas exceeding 2,000 m²/g [37], which is crucial for achieving high capacitance in supercapacitors and high capacity in batteries.
The BET theory extends the Langmuir model (for monolayer adsorption) to multilayer adsorption, describing the relationship between the amount of gas adsorbed and its relative pressure at a constant temperature (the adsorption isotherm) [37]. The core BET equation (Equation 1) is:
[ \frac{P/P0}{X(1-P/P0)} = \frac{1}{XmC} + \frac{C-1}{XmC}(P/P_0) ]
where:
- ( P/P_0 ) is the relative pressure of the adsorbate gas
- ( X ) is the volume of gas adsorbed at relative pressure ( P/P_0 )
- ( X_m ) is the monolayer capacity (the volume of gas required to form a single molecular layer)
- ( C ) is the BET constant related to the heat of adsorption
The linear form of this equation is plotted with ( \frac{P/P0}{X(1-P/P0)} ) on the y-axis versus ( P/P0 ) on the x-axis. The monolayer capacity ( Xm ) is calculated from the slope and intercept of the linear region, typically found between ( P/P_0 ) of 0.05 to 0.35 when using nitrogen as the adsorbate [37]. The specific surface area ( SA ) is then determined using Equation 2:
[ SA = \frac{X_m N \sigma}{M} ]
where:
- ( N ) is Avogadro's number
- ( \sigma ) is the cross-sectional area of the adsorbate molecule (e.g., 0.162 nm² for nitrogen at 77 K)
- ( M ) is the molar volume of the adsorbate gas
Table 1: Key Parameters in BET Surface Area Analysis for Porous Carbons
| Parameter | Description | Typical Values/Considerations for Porous Carbons |
|---|---|---|
| Adsorbate Gas | Gas molecules that physically adsorb onto the solid surface | N₂ at 77 K is most common; CO₂ at 273 K for ultramicropores |
| Monolayer Capacity (Xₘ) | Volume of gas required to form a complete monolayer | Directly proportional to the total surface area |
| BET Constant (C) | Related to the magnitude of the adsorbent-adsorbate interactions | High C value indicates strong interaction (microporous materials) |
| Linear Range | Relative pressure range where BET equation is valid | Typically P/P₀ = 0.05-0.35 for N₂ on most carbons |
| Cross-sectional Area (σ) | Area occupied by a single adsorbate molecule on the surface | N₂: 0.162 nm²; CO₂: 0.195 nm² (values depend on method) |
Experimental Protocol for Porous Carbon Electrodes
Sample Preparation:
- Degassing: Pre-treat the porous carbon sample (typically 50-200 mg) under vacuum or flowing inert gas at elevated temperature (e.g., 150-300°C for carbons) for several hours (2-12 hours) to remove any pre-adsorbed contaminants, moisture, and volatile species that could occupy adsorption sites.
- Mass Determination: Precisely weigh the clean, dry sample tube before and after sample introduction to determine the exact mass of the degassed sample.
Measurement Procedure:
- Cryogenic Conditions: Immerse the sample tube in a cryogenic bath (typically liquid nitrogen at 77 K for N₂ adsorption) to maintain constant isothermal conditions during analysis.
- Gas Introduction: Systematically introduce known amounts of the adsorbate gas (N₂) into the sample chamber while monitoring the equilibrium pressure.
- Adsorption Isotherm: Measure the quantity of gas adsorbed at each relative pressure step from low (near vacuum) to high (saturation) P/P₀.
- Desorption Isotherm: Reverse the process by gradually decreasing the pressure and measuring gas desorbed, which provides information on pore geometry through hysteresis analysis.
Data Analysis:
- BET Plot: Plot the adsorption data according to the linear BET transformation in the relative pressure range of 0.05-0.35.
- Monolayer Capacity: Calculate ( Xm ) from the slope (( s )) and intercept (( i )) of the linear fit: ( Xm = 1/(s + i) ).
- Surface Area Calculation: Compute the specific surface area using the cross-sectional area of the adsorbate molecule and Avogadro's number.
It is crucial to note recent research highlighting reproducibility challenges in BET area determination, emphasizing the need for standardized calculation approaches, particularly for nanoporous materials [38].
Application to Ion Adsorption in Porous Carbon Electrodes
For ion adsorption in porous carbon electrodes, BET analysis provides the foundational metric of accessible surface area. However, researchers must recognize that the BET surface area represents the total area accessible to gas molecules, which may differ from the electrochemically active surface area accessible to solvated ions in an electrolyte. The pore size distribution derived from the full adsorption isotherm provides additional critical information, as the relationship between pore size and ion size significantly influences adsorption capacity and kinetics.
Recent studies on zinc-ion hybrid supercapacitors demonstrate the critical importance of matching pore channels to hydrated ion dimensions. Wood-derived porous carbon with hierarchical pore structure was synthesized, and pore diameters were successfully enlarged from 0.54 nm to 0.71 nm and 1.13 nm to better accommodate [Zn·(H₂O)₆]²⁺ complexes with a diameter of ∼0.86 nm [17]. This deliberate pore size modulation resulted in a significantly enhanced specific capacitance of 412.76 F/g and excellent cycling stability (93.55% capacity retention after 20,000 cycles) [17]. Similarly, research on sodiation mechanisms in hard carbon electrodes has identified three distinct stages: adsorption at defects and edges, intercalation between graphene layers, and nano-pore filling, with the latter being particularly important for capacity [39].
Diagram 1: BET analysis workflow for porous carbon.
FT-IR Spectroscopy
Fundamental Principles
Fourier Transform Infrared (FT-IR) spectroscopy is based on the principle that chemical bonds vibrate at characteristic frequencies when exposed to infrared radiation [40]. When the frequency of the infrared light matches the natural vibrational frequency of a molecular bond, absorption occurs, resulting in a spectrum that serves as a molecular "fingerprint" of the material. For porous carbon electrodes, FT-IR is particularly valuable for identifying surface functional groups that significantly influence ion adsorption behavior, wettability, and electrochemical reactivity.
Experimental Methodology
Sample Preparation Methods:
- Transmission Spectroscopy (TS): The traditional method where a small amount of carbon sample (1-2 mg) is thoroughly mixed with potassium bromide (KBr, 100-200 mg) and pressed into a transparent pellet under high pressure. However, this method can distort delicate structures and introduce moisture interference for porous carbons [41].
- Attenuated Total Reflectance (ATR): Requires minimal sample preparation as the porous carbon material is simply placed in direct contact with the ATR crystal. This technique is particularly suitable for opaque carbon samples and allows for non-destructive analysis [41].
- Diffuse Reflectance (DRIFTS): Effective for powdered carbon samples with minimal preparation, measuring light scattered from rough surfaces or powders.
Measurement Protocol:
- Background Scan: Collect a background spectrum without the sample to account for atmospheric CO₂, water vapor, and instrument characteristics.
- Sample Scanning: Place the prepared sample in the FT-IR spectrometer and acquire the spectrum typically over the mid-IR range (4000-400 cm⁻¹) with a resolution of 4 cm⁻¹, averaging 32-64 scans to improve signal-to-noise ratio.
- Data Processing: Apply baseline correction and atmospheric suppression algorithms to enhance spectral features.
Table 2: Key FT-IR Absorbance Bands for Porous Carbon Surface Characterization
| Wavenumber (cm⁻¹) | Functional Group | Vibration Type | Significance for Ion Adsorption |
|---|---|---|---|
| 3200-3600 | O-H stretching | Stretch | Hydrophilicity, H-bonding with electrolyte |
| 2800-3000 | C-H stretching | Stretch | Presence of alkyl chains |
| 1700-1750 | C=O stretching | Stretch | Carboxyl, quinone groups; redox activity |
| 1580-1620 | C=C stretching | Stretch | Aromatic backbone; electronic conductivity |
| 1200-1300 | C-O stretching | Stretch | Phenol, ether groups; wettability |
| 1000-1100 | C-O-C stretching | Stretch | Ether linkages |
Application to Ion Adsorption Mechanisms
FT-IR spectroscopy provides critical insights into the surface chemistry of porous carbon electrodes, which directly governs ion adsorption mechanisms. Specific functional groups identified through FT-IR analysis can facilitate ion adsorption through various mechanisms:
Carboxyl groups (-COOH) identified at ~1700 cm⁻¹ are particularly important for metal ion adsorption through ion-exchange mechanisms, as demonstrated in biomass-derived carbons where carboxyl groups were identified as the main functional groups involved in metal sorption [42]. Hydroxyl groups (-OH) detected around 3200-3600 cm⁻¹ enhance hydrophilicity and facilitate access of aqueous electrolytes to the carbon surface. Carbonyl/quinone groups (C=O) contribute to pseudocapacitance through reversible redox reactions in both aqueous and non-aqueous electrolytes.
For green-synthesized carbon materials, FT-IR verifies the presence of biomolecular capping agents that stabilize the material and influence its interaction with ions [41]. The technique can also monitor chemical changes in carbon electrodes during electrochemical cycling, such as the oxidation of surface functional groups or degradation of binders.
Scanning Electron Microscopy (SEM)
Principles and Capabilities
Scanning Electron Microscopy (SEM) provides high-resolution imaging of material surfaces by scanning with a focused beam of electrons. The interactions between electrons and atoms in the sample generate various signals that reveal information about surface topography, morphology, and composition. For porous carbon electrodes, SEM is indispensable for visualizing pore structures, particle morphology, and spatial distribution of different phases at the micro- and nanoscale.
Advanced Methodologies for Porous Carbons
Standard SEM Imaging:
- Sample Preparation: Mount a small portion of the carbon electrode on a SEM stub using conductive tape. For non-conductive carbon materials, apply a thin coating (5-20 nm) of gold, platinum, or carbon using sputter coating to prevent charging effects.
- Imaging Conditions: Operate at accelerating voltages typically between 5-15 kV for carbon materials to balance surface detail with sufficient penetration. Use working distances of 5-10 mm for optimal resolution.
Focused Ion Beam-SEM (FIB-SEM) Tomography: For three-dimensional structural quantification of porous carbon electrodes, FIB-SEM represents a powerful advanced technique. The protocol involves:
- Contrast Enhancement: Infiltrate the porous carbon with a contrasting agent. A method using Pt filling of pores from gaseous precursors has demonstrated drastically improved image contrast (42% vs 20% with conventional Si-resin impregnation) between carbon and pore phases [43].
- Serial Sectioning: Use the focused ion beam to sequentially mill away thin slices (e.g., 10-20 nm thick) of the sample.
- Image Acquisition: After each milling step, acquire a high-resolution SEM image of the newly exposed surface.
- 3D Reconstruction: Align the sequential 2D images and apply segmentation algorithms to reconstruct the three-dimensional microstructure for quantitative analysis of porosity, tortuosity, pore size distribution, and specific surface area [43].
Image Processing and Analysis: Modern SEM analysis employs sophisticated image processing and computer vision (IPCV) techniques, often using software like MATLAB, to extract quantitative morphological parameters [44]. Key analyses include:
- Porosity determination through thresholding and binarization
- Pore size distribution based on the distance from the medial axis of pores to the nearest solid boundary
- Fractal dimension calculation to quantify surface complexity
- Tortuosity analysis in both axial and radial directions
Table 3: Quantitative Structural Parameters of Porous Carbons from SEM Analysis
| Parameter | Definition | Representative Values | Influence on Ion Adsorption |
|---|---|---|---|
| Porosity (%) | Volume fraction of pores | Toyo Carbon: 15%; SGL Graphite: 17%; Super Carbon: 27% [44] | Higher porosity → more ion storage sites |
| Pore Size (nm) | Characteristic pore dimension | Average: 90 nm (range 45-134 nm) [43] | Must match hydrated ion size for optimal filling |
| Tortuosity | Complexity of pore pathways | Axial: 1.45; Radial: 1.43 [43] | Lower tortuosity → faster ion transport |
| Fractal Dimension | Surface complexity measure | Varies with material; higher = more complex | Complex surfaces enhance ion adsorption |
| Specific Surface Area (μm⁻¹) | Surface-to-volume ratio | 13 μm⁻¹ [43] | Directly correlates with adsorption capacity |
Application to Porous Carbon Electrode Structure
SEM analysis provides direct visual evidence of the hierarchical pore structure in carbon electrodes, which is critical for understanding ion adsorption behavior. For instance, research on porous graphite materials has shown how structural parameters influence functional performance in applications like aerostatic bearings [44], with similar principles applying to electrochemical systems. The fractal dimension of carbon materials, which can be determined through SEM image processing, correlates with surface complexity and ion accessibility.
Studies have demonstrated that accurate determination of fractal dimension and permeability of porous graphite materials is essential for applications requiring controlled fluid (or ion) transport [44]. The relative density of the internal structure of these materials decreases with increasing fractal dimension, affecting both mechanical and transport properties [44]. These structural insights guide the rational design of carbon electrodes with optimized pore architectures for specific ion types and operating conditions.
Diagram 2: SEM analysis workflow for porous carbon electrodes.
Elemental Analysis
Techniques and Methodologies
Elemental analysis encompasses a suite of techniques to determine the elemental composition of materials. For porous carbon electrodes, understanding elemental composition is crucial as it influences electronic conductivity, surface functionality, and electrochemical stability.
Key Techniques:
- CHNS/O Analysis: Combustion analysis that determines carbon, hydrogen, nitrogen, sulfur, and oxygen content by complete combustion of the sample and quantification of resulting gases.
- X-ray Photoelectron Spectroscopy (XPS): Provides both elemental composition and chemical state information for surface layers (top 1-10 nm), which is particularly relevant for ion adsorption processes.
- Energy-Dispersive X-ray Spectroscopy (EDS/EDX): Often coupled with SEM to provide elemental mapping and spatial distribution of elements within the carbon matrix.
- Inductively Coupled Plasma (ICP) Techniques: Determine trace metal content in carbon materials, which can affect electrochemical behavior.
Application to Ion Adsorption in Carbon Electrodes
Elemental composition significantly influences the ion adsorption behavior of porous carbon electrodes. Research on biomass-derived carbons has revealed that materials with higher carbon, nitrogen, and hydrogen content generally exhibit lower metal sorption capacity, suggesting that heteroatom doping must be optimized rather than maximized [42]. The presence of specific heteroatoms like oxygen, nitrogen, and sulfur creates surface functional groups that enhance ion adsorption through various mechanisms:
Nitrogen-containing groups (pyridinic, pyrrolic, quaternary) improve electronic conductivity and introduce pseudocapacitive behavior through faradaic reactions. Oxygen functional groups (carboxyl, hydroxyl, carbonyl) enhance hydrophilicity and enable ion-exchange mechanisms, with studies showing that carboxyl groups are particularly relevant for metal ion sorption [42]. Sulfur and phosphorus doping can alter the electronic structure of carbon and provide specific interactions with certain ions.
Ion-exchange has been identified as a primary metal sorption mechanism in many carbonaceous materials, with the release of K⁺ and Ca²⁺ ions during metal sorption indicating the occurrence of cation exchange processes [42]. This understanding guides the design of carbon electrodes with tailored surface chemistry for specific ion adsorption applications.
Integrated Characterization Approach
Correlative Analysis Framework
A comprehensive understanding of ion adsorption in porous carbon electrodes requires the integration of data from all characterization techniques. The following diagram illustrates how these techniques interrelate to provide a complete picture of structure-property relationships:
Diagram 3: Integrated characterization for ion adsorption analysis.
Research Reagent Solutions for Porous Carbon Characterization
Table 4: Essential Research Reagents and Materials for Porous Carbon Electrode Characterization
| Reagent/Material | Function | Application Examples |
|---|---|---|
| High-Purity N₂ Gas (99.999%) | Adsorbate for BET analysis | Surface area and pore size distribution measurements [37] |
| Liquid N₂ | Cryogenic coolant for BET | Maintaining 77 K isothermal conditions during gas adsorption [45] |
| KBr (Potassium Bromide) | IR-transparent matrix | FT-IR sample preparation via pellet method [41] |
| Gold/Pt Sputtering Target | Conductive coating | SEM sample preparation for non-conductive carbons [43] |
| Pt Gaseous Precursors | Contrast enhancement agent | FIB-SEM tomography for 3D structural quantification [43] |
| Helium/Hydrogen Carrier Gas | BET analysis carrier | Transport gas in flow-type BET instruments [45] |
| Standard Reference Materials | BET calibration | Certified surface area standards for method validation [38] |
The comprehensive characterization of porous carbon electrodes for ion adsorption applications requires a multifaceted approach integrating BET surface area analysis, FT-IR spectroscopy, SEM, and elemental analysis. Each technique provides unique and complementary information about the physical and chemical properties that govern ion adsorption behavior. BET analysis quantifies the accessible surface area and pore structure, FT-IR identifies critical surface functional groups, SEM reveals morphological features and 3D architecture, while elemental analysis determines composition and heteroatom doping. Together, these techniques form a powerful toolkit for researchers developing advanced carbon electrodes for energy storage, environmental remediation, and catalytic applications. By applying these characterization methods in an integrated framework, scientists can establish robust structure-property relationships that guide the rational design of next-generation porous carbon materials with optimized performance for specific ion adsorption applications.
The evaluation of ion removal efficiency is a fundamental aspect of developing advanced porous carbon electrodes for applications ranging from energy storage to water purification. Batch adsorption experiments represent a critical methodology for systematically investigating the capacity, kinetics, and mechanisms of ion uptake by porous carbon materials. Within the broader context of ion adsorption in porous carbon electrodes research, these experiments provide essential data on electrosorption capabilities, ion transport phenomena, and interface behaviors that directly influence the performance of supercapacitors and adsorption-based separation systems.
The significance of batch adsorption studies has been particularly highlighted by recent investigations into complex ion behavior at carbon interfaces. Research has revealed that potassium ions, for instance, can exist in five distinct desolvation states ([K(H₂O)₀₋₄]⁺) when interacting with porous carbon structures, each with unique thermodynamic and kinetic properties that ultimately govern electric double-layer capacitance [15]. Understanding these fundamental interactions through standardized batch experimentation provides the scientific foundation for designing next-generation carbon materials with enhanced ion adsorption characteristics, thereby bridging the gap between fundamental molecular-level processes and macroscopic electrochemical performance.
Fundamental Principles of Batch Adsorption
Batch adsorption experiments investigate the distribution of ions between a solid adsorbent phase and a liquid solution phase at equilibrium conditions. For porous carbon electrodes, this process is governed by multiple interfacial phenomena, including electrostatic interactions, ion solvation/desolvation, and pore size effects. The experimental approach allows researchers to quantify key parameters that define adsorption performance, including equilibrium capacity, removal efficiency, and adsorption kinetics.
The ion removal mechanism in porous carbon materials involves a complex sequence of steps beginning with bulk diffusion of ions to the carbon surface, followed by boundary layer diffusion, and ultimately, adsorption at active sites within the porous structure. Recent advances have demonstrated that the desolvation state of ions within carbon nanopores significantly influences the overall adsorption energy and capacity, with partially desolvated ions often exhibiting optimal adsorption characteristics [15]. This fundamental understanding enables more precise engineering of carbon materials for specific ion adsorption applications.
Experimental Design and Setup
Research Reagent Solutions and Essential Materials
| Material/Reagent | Specification | Primary Function |
|---|---|---|
| Porous Carbon Adsorbent | High surface area (>500 m²/g), controlled pore size distribution | Primary adsorption material; provides active sites for ion uptake |
| Target Ion Solution | Standard solutions with precise concentrations (e.g., 10-50 ppm for screening) | Simulates contaminated water; enables adsorption isotherm construction |
| pH Adjustment Solutions | HCl and NaOH solutions (0.1-1.0 M) | Controls solution acidity; influences adsorbent surface charge and ion speciation |
| Electrolyte Background | Inert salts (e.g., NaCl, Na₂SO₄) at controlled ionic strength | Maintains constant ionic strength; simulates real water matrices |
| Characterization Reagents | Various chemicals for surface modification and analysis | Enhances adsorbent properties; enables mechanism investigation |
Equipment and Instrumentation
A properly equipped laboratory for batch adsorption experiments should contain the following essential equipment: mechanical shaker or orbital incubator shaker (for constant agitation); centrifuge (for phase separation after adsorption); pH meter (for precise pH measurement and adjustment); analytical balance (for accurate mass measurements of adsorbent); atomic absorption spectrometer (AAS) or inductively coupled plasma optical emission spectrometer (ICP-OES) (for ion concentration measurement); and temperature-controlled water bath (for isothermal studies). For advanced characterization of porous carbon materials post-adsorption, Brunauer-Emmett-Teller (BET) surface area analyzer, Fourier-transform infrared spectroscopy (FTIR), and scanning electron microscope with energy-dispersive X-ray spectroscopy (SEM-EDX) are recommended.
Experimental Parameters and Optimization
The efficiency of ion removal in batch adsorption systems is influenced by multiple interconnected parameters that must be systematically optimized to evaluate material performance accurately. The table below summarizes key parameters, their typical ranges, and optimization approaches based on current research practices.
Table 2: Key Experimental Parameters in Batch Adsorption Studies
| Parameter | Typical Range | Effect on Adsorption | Optimization Approach |
|---|---|---|---|
| Solution pH | 2-10 [46] [47] | Affects surface charge of adsorbent and ion speciation | Evaluate removal efficiency across pH range; select optimum |
| Adsorbent Dosage | 0.2-1.0 g/100 mL [46] | Increases available adsorption sites; may cause particle aggregation | Determine minimum dosage for maximum efficiency |
| Initial Ion Concentration | 10-50 ppm (screening) [46]; up to 600 mg/L [47] | Influences driving force for adsorption; tests capacity | Use environmentally relevant concentrations for application |
| Contact Time | 5-75 min [46]; up to 120 min [47] | Determines kinetic profile and equilibrium time | Sample at regular intervals until equilibrium reached |
| Temperature | 25-90°C [46] | Affects adsorption thermodynamics and kinetics | Study multiple temperatures for thermodynamic parameters |
| Agitation Speed | 100-200 rpm | Influences external boundary layer diffusion | Maintain constant, sufficient speed to suspend particles |
Parameter Optimization Strategies
Solution pH is arguably the most critical parameter as it governs both the surface charge of the porous carbon adsorbent and the chemical speciation of target ions. For metal cation removal, adsorption typically increases as pH rises due to decreased competition between H⁺ ions and the target cations for adsorption sites [46]. The point of zero charge (PZC) of the carbon material determines the pH at which its surface becomes negatively charged and favorable for cation adsorption. Optimal pH conditions must be determined experimentally for each adsorbent-ion system.
Contact time experiments reveal the kinetic behavior of the adsorption process and the time required to reach equilibrium. Studies on modified diatomite for lead ion removal demonstrated that 45 minutes was sufficient to achieve equilibrium, while chitosan-sodium alginate-halloysite nanotube composites for dye removal reached equilibrium within 30-70 minutes depending on composition [46] [47]. These temporal profiles are essential for designing continuous flow systems and understanding the practical applicability of the adsorbent material.
Core Experimental Protocol
Adsorbent Preparation and Characterization
Begin with comprehensive characterization of the porous carbon material prior to adsorption experiments. Determine the specific surface area using BET analysis, with values for effective adsorbents typically ranging from 22.39–34.83 m²/g for modified diatomite to much higher values for advanced porous carbons [46]. Analyze surface functional groups using FTIR spectroscopy and examine morphological features using SEM. Determine the point of zero charge (PZC) using the solid addition method, as this parameter critically influences pH optimization.
For modified adsorbents, carefully execute synthesis protocols. For example, acid-modified diatomite preparation involves treatment with sulfuric acid (2-10 M concentration range) at elevated temperatures (50-90°C) for specified durations (4-12 hours) with controlled solid-to-liquid ratios (50-250 g/L) [46]. Similarly, composite materials like chitosan/sodium alginate/halloysite nanotube (CSAH) composites require precise control of component ratios (5-30% HNT content) during synthesis [47]. These preparation parameters significantly influence the final adsorbent properties and must be meticulously documented.
Batch Adsorption Procedure
The following workflow outlines the standard procedure for conducting batch adsorption experiments:
Figure 1: Batch Adsorption Experimental Workflow
Solution Preparation: Prepare stock solutions of the target ions at precise concentrations (e.g., 1000 mg/L) using analytical grade salts and deionized water. Dilute to desired working concentrations (typically 10-50 mg/L for initial screening studies) [46].
pH Adjustment: Adjust the pH of the solutions using 0.1 M HCl or NaOH solutions, monitoring with a calibrated pH meter. The pH should be optimized based on preliminary experiments, with neutral pH (7.0) often providing effective results for cation removal [46].
Adsorbent Addition: Accurately weigh predetermined masses of porous carbon adsorbent (typically 0.2-1.0 g per 100 mL solution) [46] and add to the solution. Include control samples without adsorbent to account for any container adsorption or ion precipitation.
Agitation and Sampling: Agitate the mixtures at constant speed (100-200 rpm) in a temperature-controlled environment. Withdraw samples at predetermined time intervals (5, 15, 30, 45, 60, 75 minutes for kinetic studies) [46].
Separation: Centrifuge samples promptly (or filter through 0.45 μm membrane filters) to separate the adsorbent from the solution.
Analysis: Measure the residual ion concentration in the supernatant using appropriate analytical techniques (AAS, ICP-OES, or UV-Vis spectrophotometry for colored ions).
All experiments should be conducted in duplicate or triplicate to ensure data reproducibility, with appropriate blank corrections applied to all measurements.
Data Analysis and Modeling
Efficiency and Capacity Calculations
The adsorption performance is quantified using two primary parameters: removal efficiency and adsorption capacity. The removal efficiency (% R) is calculated as:
% R = (C₀ - Cₑ)/C₀ × 100
where C₀ is the initial ion concentration (mg/L) and Cₑ is the equilibrium ion concentration (mg/L).
The equilibrium adsorption capacity (Qₑ, mg/g) is calculated as:
Qₑ = (C₀ - Cₑ)V/m
where V is the solution volume (L) and m is the mass of adsorbent (g).
These calculations provide the fundamental metrics for comparing different adsorbent materials and optimization conditions.
Adsorption Isotherm Modeling
Adsorption isotherms describe the equilibrium relationship between the concentration of ions in solution and the amount adsorbed on the solid phase at constant temperature. The table below summarizes the most relevant isotherm models for porous carbon ion adsorption systems:
Table 3: Adsorption Isotherm Models for Ion Removal Data Analysis
| Model | Equation | Parameters | Application Context |
|---|---|---|---|
| Langmuir | Qₑ = (Qₘₐₓ·Kₗ·Cₑ)/(1 + Kₗ·Cₑ) | Qₘₐₓ: maximum capacity; Kₗ: affinity constant | Monolayer adsorption on homogeneous surfaces [46] |
| Freundlich | Qₑ = Kꜰ·Cₑ¹/ⁿ | Kꜰ: capacity indicator; n: intensity parameter | Heterogeneous surfaces with multilayer adsorption |
| Sips | Qₑ = (Qₘₐₓ·Kₛ·Cₑⁿₛ)/(1 + Kₛ·Cₑⁿₛ) | Qₘₐₓ: maximum capacity; Kₛ: Sips constant; nₛ: heterogeneity parameter | Combines Langmuir and Freundlich; heterogeneous surfaces [46] |
| Toth | Qₑ = Qₘₐₓ·Cₑ/(Kₜ + Cₑ)¹/ᵗ | Kₜ: Toth constant; t: heterogeneity parameter | Asymmetric heterogeneity; porous materials [47] |
Model selection should be based on both statistical criteria (R², error analysis) and physical plausibility. For porous carbon materials, the Sips and Toth models often provide excellent fits due to their ability to account for surface heterogeneity [46] [47]. The maximum adsorption capacity (Qₘₐₓ) obtained from these models provides a standardized metric for comparing different adsorbent materials.
Adsorption Kinetic Modeling
Kinetic analysis reveals the temporal evolution of the adsorption process and potential rate-limiting steps. The primary kinetic models applied to ion adsorption on porous carbons include:
Table 4: Adsorption Kinetic Models for Ion Removal Data Analysis
| Model | Equation | Parameters | Application Context |
|---|---|---|---|
| Pseudo-First-Order | Qₜ = Qₑ(1 - e⁻ᵏ¹ᵗ) | k₁: rate constant; Qₑ: equilibrium capacity | Physical adsorption; diffusion-limited systems |
| Pseudo-Second-Order | Qₜ = (k₂·Qₑ²·t)/(1 + k₂·Qₑ·t) | k₂: rate constant; Qₑ: equilibrium capacity | Chemisorption; rate-limited by surface sites [46] [47] |
| Intraparticle Diffusion | Qₜ = kᵢ·t¹/² + C | kᵢ: diffusion rate constant; C: boundary layer thickness | Pore diffusion mechanisms; identifies rate-limiting steps [46] |
The pseudo-second-order model has been found to accurately describe adsorption kinetics for various ion-porous carbon systems, suggesting that the rate-limiting step often involves chemisorption mechanisms [46] [47]. Intraparticle diffusion modeling can further elucidate whether pore diffusion controls the overall adsorption rate, which is particularly relevant for hierarchical porous carbon structures.
Advanced Considerations for Porous Carbon Electrodes
When evaluating ion removal efficiency specifically for porous carbon electrodes, several advanced considerations become paramount. The ion desolvation process at the electrode-electrolyte interface significantly influences the electric double-layer formation and subsequent adsorption capacity. Recent studies have identified that potassium ions can exist in five distinct desolvation states ([K(H₂O)₀₋₄]⁺) within porous carbon structures, each with unique thermodynamic and kinetic properties [15]. Understanding these states is crucial for designing carbon materials with optimized pore sizes and surface chemistry.
The incorporation of oxygen functional groups and other heteroatoms into carbon matrices can dramatically enhance ion adsorption through multiple mechanisms, including improved wettability, introduction of specific binding sites, and modulation of electronic properties. A comprehensive design strategy based on dual thermodynamic-kinetic optimization principles has been shown to identify appropriate types and concentration windows of oxygen groups that synergize with specific ion desolvation states, leading to significantly enhanced capacitance and adsorption performance [15].
Furthermore, the structural disorder in carbon materials, conceptualized through "entropy-driven" design principles including unit entropy, ring entropy, and element entropy, can create diverse adsorption sites capable of accommodating various ion species and solvation states [13]. This approach enables the development of high-entropy carbon materials with exceptional ion adsorption capabilities and electrochemical performance.
Quality Control and Method Validation
Robust batch adsorption experiments require rigorous quality control and method validation to ensure data reliability and reproducibility. Key quality assurance measures include: blank corrections to account for any container adsorption or experimental artifacts; replicate measurements (minimum of duplicates) to assess precision; control experiments without adsorbent to monitor potential ion precipitation or degradation; standard reference materials when available to verify analytical accuracy; and mass balance calculations to confirm recovery efficiency.
Method validation should establish linear calibration ranges for analytical instruments, detection and quantification limits for target ions, precision estimates through repeated measurements, and reproducibility across different experimental batches. Documentation of all experimental parameters, including temperature fluctuations, solution preparation records, and instrument calibration data, is essential for experimental integrity and future reproducibility.
For porous carbon materials specifically, additional characterization of adsorbent properties both before and after adsorption experiments provides valuable insights into adsorption mechanisms and potential material changes during the process. Comparison of BET surface area, pore size distribution, and surface chemistry pre- and post-adsorption can reveal pore blockage, surface oxidation, or other modifications that may influence long-term performance.
The intrinsic property of porous carbon electrodes to adsorb ions, which forms the fundamental basis of operation for supercapacitors and capacitive deionization (CDI), is being strategically leveraged to address one of the most pressing global environmental challenges: water contamination by heavy metals. This whitepaper synthesizes current research demonstrating how the rational design of carbon electrodes—through heteroatom doping, surface functionalization, and structural engineering—can transform them into highly efficient, selective, and reusable platforms for purifying water. By framing these advancements within the broader context of ion adsorption research, we provide a technical guide for scientists developing next-generation environmental remediation technologies that bridge the gap between energy storage and water security.
The electric double layer (EDL) formation at the electrode-electrolyte interface, a well-established concept in electrochemical energy storage, is fundamentally an ion adsorption process. In capacitive deionization (CDI), this principle is directly applied to water treatment: when a pair of porous carbon electrodes is charged, anions and cations from the water are electrosorbed into the respective electrodes, thereby desalinating the water stream [48]. The process is reversed during discharge, regenerating the electrodes and releasing the concentrated ions.
This mechanism is exceptionally suited for the removal of toxic heavy metal ions such as Pb²⁺, Cd²⁺, and Cu²⁺ from wastewater. The performance of a CDI system is explicitly governed by the physicochemical properties of the electrode material, including its specific surface area, pore structure, surface functional groups, and electrical conductivity [48]. Consequently, the extensive research focus on enhancing ion adsorption in porous carbons for supercapacitors directly translates to improved efficacy in water purification. The following sections detail the material design strategies and experimental evidence underpinning this application.
Material Design Strategies for Enhanced Metal Ion Adsorption
The adsorption capacity and selectivity of porous carbon electrodes for heavy metals are enhanced through several key material design strategies.
Incorporation of Transition Metal Oxides
The incorporation of pseudocapacitive transition metal oxides onto carbon scaffolds introduces Faradaic reactions, significantly boosting ion storage capacity beyond the limits of physical EDL formation.
Cobalt Oxide (Co₃O₄) Nanocomposites: A composite of Co₃O₄ nanoparticles on palm kernel shell-derived activated carbon (PKSAC) was synthesized via a hydrothermal technique at 160°C. This material exhibited a mesoporous structure with a specific surface area of 1200 m²/g and delivered a specific capacitance of 414.3 F/g, outperforming the pristine PKSAC (365.4 F/g). When configured in an asymmetric CDI cell (PKSAC//Co₃O₄-PKSAC), it achieved remarkable electrosorption capacities of 56.32 mg/g for Pb²⁺ and 45.71 mg/g for Cd²⁺, compared to 23.21 mg/g for NaCl, demonstrating a pronounced selectivity for multivalent heavy metal cations [48].
Copper-Doped Carbon (Cu-C) from Waste Valorization: Research has shown a complete solution where Cu(II) is first removed from wastewater via flocculation. The resulting Cu-laden sludge is then pyrolyzed to produce Cu-doped porous carbon. This material functions as an excellent electrode for supercapacitors, achieving a high specific capacity of 389.9 F/g at 1 A/g, while simultaneously immobilizing the toxic heavy metal within a stable carbon matrix, preventing secondary environmental release [49].
Heteroatom Doping and Surface Functionalization
Introducing heteroatoms like nitrogen (N) and oxygen (O) into the carbon lattice alters its electronic structure and surface chemistry, enhancing hydrophilicity and providing specific binding sites for metal ions.
Nitrogen/Oxygen Co-Doping: A nitrogen/oxygen-codoped dense porous carbon (NDPC) was developed using a co-chemical welding strategy with carbon dots and melamine. The optimized material possessed a high compaction density of 1.19 g cm⁻³. Density functional theory (DFT) calculations confirmed that the N/O co-doping enhances K⁺ adsorption energy through optimized electronic structure, a principle that extends to the stronger adsorption of heavy metal cations. The electrode achieved volumetric and gravimetric specific capacitances of 373.6 F cm⁻³ and 314 F g⁻¹, respectively [18].
Graft Copolymerization with Acrylic Acid: Surface functional groups were dramatically increased by grafting acrylic acid onto teak wood-derived hydrochar (TH). This process enhanced the density of carboxyl (-COOH) and hydroxyl (-OH) groups, which are key for metal ion complexation. Post-grafting, the maximum adsorption capacity for Pb²⁺ surged from 116 mg/g to 294 mg/g. The adsorption isotherms followed the Langmuir model, confirming a chemisorption mechanism dominated by ion exchange and surface complexation [50].
Hierarchical and Sponge-Like Porous Architectures
A hierarchical pore structure, integrating macropores, mesopores, and micropores, is critical for facilitating ion transport and maximizing accessible adsorption sites.
- Sponge-Like Edge Structures: A highly sensitive electrochemical heavy metal sensor was developed based on hierarchical nanoporous carbon electrodes with sponge-like edges fabricated via C-MEMS and O₂ plasma etching. The hierarchical architecture provided large active sites and fast electron transfer, enabling the efficient formation of dense heavy metal alloy particles during the preconcentration step. This design allowed for the detection of Cd(II) and Pb(II) at concentrations as low as 0.41 and 0.7 μg L⁻¹, respectively [51].
Table 1: Performance Summary of Engineered Carbon Materials in Heavy Metal Removal and Related Electrochemical Performance.
| Material | Modification Strategy | Target Pollutant | Key Performance Metric | Related Capacitance |
|---|---|---|---|---|
| Co₃O₄-PKSAC [48] | Transition metal oxide coating | Pb²⁺, Cd²⁺, NaCl | 56.32, 45.71, and 23.21 mg/g electrosorption capacity | 414.3 F/g |
| Cu-Doped Carbon [49] | In-situ metal doping from sludge | Cu²⁺ (removed during synthesis) | >99% Cu(II) removal efficiency | 389.9 F/g |
| N/O-Doped Carbon (NDPC) [18] | Heteroatom doping & compaction | K⁺ (model ion) | Enhanced K⁺ adsorption energy (DFT) | 314 F/g (Gravimetric) |
| Acrylic Acid-Grafted Hydrochar [50] | Graft copolymerization | Pb²⁺, Cu²⁺, Cd²⁺ | 294, 164, and 170 mg/g adsorption capacity | Not Specified |
| Hierarchical Nanoporous Carbon [51] | Sponge-like edge structures | Cd²⁺, Pb²⁺ | Detection limits of 0.41 and 0.7 μg L⁻¹ | Not Specified |
Experimental Protocols and Methodologies
Synthesis of Transition Metal Oxide-Carbon Nanocomposites
Protocol: Hydrothermal Synthesis of Co₃O₄-PKSAC [48]
- Precursor Preparation: Begin with activated carbon (AC) derived from palm kernel shells (PKS), produced via carbonization at 800°C using CO₂ and ZnCl₂ as activating agents.
- Solution Preparation: Dissolve Cobalt nitrate (Co(NO₃)₂·6H₂O) in deionized water to form a clear solution.
- Mixing: Immerse the PKSAC powder in the cobalt nitrate solution under continuous stirring to ensure homogeneous dispersion.
- Hydrothermal Reaction: Transfer the mixture to a Teflon-lined stainless-steel autoclave. Seal and maintain the autoclave at 160°C for several hours (specific time to be optimized) to facilitate the growth of Co₃O₄ nanoparticles on the carbon surface.
- Washing and Drying: After the reaction, cool the autoclave to room temperature. Collect the solid product by filtration and wash repeatedly with deionized water and ethanol to remove any unreacted precursors. Dry the final Co₃O₄-PKSAC nanocomposite in an oven at 60-80°C overnight.
Capacitive Deionization (CDI) Testing for Heavy Metal Removal
Protocol: Electrosorption Capacity Measurement [48]
- Electrode Fabrication: Mix the active material (e.g., PKSAC or Co₃O₄-PKSAC), a conductive agent (e.g., carbon black), and a binder (e.g., PVDF) in a mass ratio of 80:10:10 in a suitable solvent (e.g., N-Methyl-2-pyrrolidone, NMP) to form a slurry. Coat the slurry onto a current collector (e.g., graphite sheet) and dry thoroughly.
- Cell Assembly: Construct an asymmetric CDI cell with one electrode serving as the anode and the other as the cathode, separated by a spacer or membrane.
- Solution Preparation: Prepare a synthetic wastewater solution containing the target heavy metal ions (e.g., Pb²⁺ from Pb(NO₃)₂, Cd²⁺ from Cd(NO₃)₂·4H₂O) at a known initial concentration (e.g., 750 mg/L).
- CDI Operation: Circulate the solution through the CDI cell while applying a constant voltage (e.g., 1.2 V) across the electrodes using a power source. Monitor the concentration of the effluent over time using techniques like Inductively Coupled Plasma Optical Emission Spectroscopy (ICP-OES) or Atomic Absorption Spectroscopy (AAS).
- Data Analysis: Calculate the electrosorption capacity (Q, in mg/g) using the formula: ( Q = (C0 - Ce) \times V / m ) where ( C0 ) and ( Ce ) are the initial and equilibrium concentrations (mg/L), respectively, ( V ) is the volume of the solution (L), and ( m ) is the total mass of the active material on both electrodes (g).
Adsorption Isotherm Modeling
Protocol: Fitting Equilibrium Data [52] [53]
- Batch Experiments: Conduct a series of batch adsorption experiments by varying the initial concentration of the heavy metal ion while keeping the adsorbent dose, pH, temperature, and contact time constant. The contact time must be sufficient to reach equilibrium.
- Equilibrium Concentration Measurement: After reaching equilibrium, separate the adsorbent from the solution and measure the final concentration of the heavy metal ion, ( C_e ) (mg/L).
- Capacity Calculation: Calculate the adsorption capacity at equilibrium, ( q_e ) (mg/g), for each initial concentration using the formula above.
- Model Fitting: Fit the (( Ce ), ( qe )) data pairs to various isotherm models. The Langmuir model (assuming monolayer adsorption on a homogeneous surface) and Freundlich model (assuming multilayer adsorption on a heterogeneous surface) are most common.
- Langmuir Isotherm: ( qe = (qm \cdot KL \cdot Ce) / (1 + KL \cdot Ce) ) where ( qm ) is the maximum monolayer adsorption capacity (mg/g) and ( KL ) is the Langmuir constant (L/mg).
- Freundlich Isotherm: ( qe = KF \cdot Ce^{1/n} ) where ( KF ) and ( n ) are Freundlich constants related to adsorption capacity and intensity.
- Model Selection: Use nonlinear regression over linearization for more accurate parameter estimation. Compare the goodness-of-fit for different models using error functions like R² (coefficient of determination). The Langmuir model is frequently reported as the best fit for heavy metal adsorption, indicating a dominant chemisorption mechanism [52] [50].
Visualization of Workflows and Mechanisms
The following diagrams illustrate the core concepts, synthesis pathways, and operational mechanisms discussed in this whitepaper.
Ion Adsorption Mechanism in Porous Carbon
Diagram 1: Ion adsorption mechanism during electrode charging, showing heavy metal ion capture.
Material Synthesis and CDI Process
Diagram 2: Integrated workflow from biomass to water treatment via CDI.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 2: Key reagents, materials, and equipment for researching carbon-based heavy metal adsorption.
| Category/Item | Specific Examples | Function/Application |
|---|---|---|
| Carbon Precursors | Palm Kernel Shells [48], Teak Wood Waste [50], Coal Liquefaction Residue [18] | Sustainable, low-cost raw materials for producing porous carbons with inherent heteroatoms. |
| Activating Agents | ZnCl₂ [48], KOH [18] | Chemical activators that create and tune porosity during pyrolysis. |
| Doping/Functionalization Agents | Melamine (N-source) [18], Acrylic Acid (grafting) [50], Cobalt Nitrate (Metal oxide) [48] | Introduce heteroatoms or surface functional groups to enhance chemisorption and pseudocapacitance. |
| Heavy Metal Salts | Pb(NO₃)₂, Cd(NO₃)₂·4H₂O, CuCl₂·2H₂O [48] [49] [50] | Used to prepare synthetic wastewater for controlled adsorption experiments. |
| Electrode Fabrication | Conductive Carbon Black, Polyvinylidene Fluoride (PVDF) binder, N-Methyl-2-pyrrolidone (NMP) solvent [48] | Components for preparing stable electrode slurries and coating current collectors. |
| Analytical Instruments | ICP-OES/AAS [48], BET Surface Area Analyzer [48] [18], XPS, FTIR [50] | For quantifying heavy metal concentration, characterizing material porosity, and analyzing surface chemistry. |
The convergence of materials science for electrochemical energy storage and environmental engineering has given rise to powerful, carbon-based technologies for water remediation. The research summarized herein unequivocally demonstrates that porous carbon electrodes, when engineered with specific functionalities and architectures, transcend their traditional role in energy storage to become highly effective platforms for the selective removal of toxic heavy metals from water. The continued refinement of these materials—focusing on selectivity, stability, and scalability—holds the promise of delivering energy-efficient, cost-effective, and sustainable solutions to the global challenge of water pollution.
Strategies for Enhancing Adsorption Capacity and Kinetics
Optimizing Pyrolysis Temperature and Activator Ratios for Maximum Performance
The pursuit of advanced energy storage solutions has positioned porous carbon materials as a cornerstone of modern electrochemical research. The performance of these materials, particularly in applications like supercapacitors, is intrinsically linked to their synthesis parameters. The optimization of pyrolysis temperature and activator ratios is paramount for developing porous carbon electrodes with superior ion adsorption capabilities. This whitepaper provides a technical guide for researchers, consolidating current experimental data and protocols to navigate the complex relationship between synthesis conditions and the resulting electrochemical performance of porous carbons.
The Interplay of Pyrolysis Temperature and Carbon Properties
Pyrolysis temperature is a critical determinant of the structural and chemical properties of porous carbon. It directly influences the degree of carbonization, pore evolution, surface functionality, and ultimately, the electrochemical ion adsorption capacity.
Structural and Physicochemical Transformations
The thermal environment during pyrolysis dictates the formation of the carbon matrix. Higher temperatures generally promote the development of graphitic domains and enhance electrical conductivity, which is crucial for efficient charge transfer in electrodes [54]. However, an intricate balance must be struck, as excessive temperatures can lead to pore collapse and a reduction in functional groups that contribute to pseudocapacitance.
A study on cedar wood-derived biochar systematically investigated this balance, revealing an optimal pyrolysis temperature of 900°C [55]. At this point, the biochar exhibited a favorable combination of electrical conductivity, hydrophobicity, and porosity. Biochar produced at lower temperatures (800°C) lacked sufficient conductivity, while at higher temperatures (1000°C and 1100°C), the structural integrity was compromised, leading to diminished performance [55].
Influence on Pore Architecture and Ion Adsorption
The pyrolysis temperature also governs the pore size distribution, which is a critical factor for ion adsorption. A hierarchical pore structure, containing a mix of micropores (<2 nm), mesopores (2-50 nm), and macropores (>50 nm), is often ideal. Micropores provide a large surface area for ion electrosorption, while mesopores act as transport channels for ions to reach the micropores [54] [18].
Research on almond shell-derived porous carbon demonstrated that high-temperature pyrolysis at 1000°C post-activation successfully created a hierarchical micro/mesoporous architecture [54]. This optimized structure was pivotal in facilitating rapid ion transport and providing abundant active sites, resulting in a high specific capacitance of 142.15 F/g and excellent rate performance [54].
Table 1: Impact of Pyrolysis Temperature on Biochar Properties and Performance
| Pyrolysis Temperature (°C) | Specific Surface Area (SSA) | Key Property Changes | Observed Electrochemical Performance |
|---|---|---|---|
| 800 | Develops | Lower conductivity, functional groups preserved | Suboptimal capacitance and rate capability [55] |
| 900 | High | Optimal conductivity & porosity balance | High capacitance and stability in cedar wood biochar [55] |
| 1000 | Very High | Enhanced graphitic domains, hierarchical pores | Superior capacitance (142 F/g) & 75% retention at 20 A/g in almond shell carbon [54] |
| 1100 | May decrease | Possible pore collapse, loss of functional groups | Declining performance due to structural degradation [55] |
Optimizing Chemical Activation and Activator Ratios
Chemical activation is a cornerstone process for developing high-surface-area porous carbons. The choice of activator and its ratio to the carbon precursor are levers for precise control over the final material's pore network and surface chemistry.
Mechanisms of Alkali Activation
Alkalis like KOH and K₂CO₃ are potent activators. Traditional understanding categorized them simply by their corrosiveness, but recent research redefines their roles based on chemical mechanisms [56].
In a mixed KOH/K₂CO₃ system:
- KOH (Strong Alkali) acts as a "Promoter": It decomposes to K₂O at lower temperatures, which aggressively attacks C-C bonds to create active sites termed "pore seeds" [56].
- K₂CO₃ (Weak Alkali) acts as a "Pathway Modifier": Its carbonate ion (CO₃²⁻) preferentially etches these pore seeds with a lower energy barrier than K₂O, leading to a gentler, more controlled formation of micropores [56].
This synergistic effect allows for tailored pore architecture. By adjusting the ratio of strong (KOH) to weak (K₂CO₃) alkali, researchers can control the microporosity. A 1:1 ratio has been shown to produce carbon with an ultra-high microporosity of 82.61% and a specific surface area of 1962.18 m² g⁻¹ [56].
One-Step vs. Two-Step Activation Protocols
The sequence of activation and pyrolysis also significantly impacts the characteristics of the final carbon material.
- Two-Step Activation: This traditional method involves initial carbonization of the biomass precursor, followed by chemical activation of the resulting biochar in a second pyrolysis step. This method typically yields carbons with higher specific surface areas. For example, orange peel activated via a two-step method (KAC-2) achieved a BET surface area of 1404 m²/g and a phenol adsorption capacity of 467 mg/g [57].
- One-Step Activation: This streamlined process involves impregnating the raw biomass with the chemical activator followed by a single pyrolysis stage. While sometimes resulting in a lower surface area (e.g., 956 m²/g for orange peel-derived carbon KAC-1), it offers a less complex and potentially more cost-effective route while still delivering strong performance (phenol adsorption of 360 mg/g) [57].
Experimental Protocols for Synthesis and Characterization
This section outlines detailed methodologies for synthesizing and evaluating high-performance porous carbons, drawing from established protocols in recent literature.
Detailed Synthesis Workflow
Protocol 1: Two-Step Chemical Activation with KOH [57]
- Precursor Preparation: Dry biomass (e.g., orange peel) at 105°C overnight. Mill and sieve to a fine powder (<0.210 mm).
- Initial Carbonization: Pyrolyze the raw biomass in a tubular reactor under a continuous N₂ flow (e.g., 400 mL/min). Heat to a moderate temperature (e.g., 500°C) at a defined heating rate (e.g., 8°C/min) and hold for a set time (e.g., 60 min). Cool to room temperature under N₂ to obtain biochar.
- Chemical Impregnation: Mix the biochar with an KOH solution at a desired impregnation ratio (e.g., 1:1 by weight). Stir for 24 hours to ensure uniformity, then dry at 110°C.
- Activation Pyrolysis: Place the impregnated biochar in a reactor and pyrolyze under N₂ flow. Heat to the high target activation temperature (e.g., 800°C) at a controlled rate, hold for the activation time (e.g., 60 min), then cool to room temperature.
- Post-processing: Wash the resulting activated carbon with HCl solution (e.g., 0.1 M) to remove inorganic residues, followed by rinsing with deionized water until neutral pH. Dry the final product.
Protocol 2: Mixed Alkali Activation for Tuned Microporosity [56]
- Precursor and Activator Mixing: Mix the carbon precursor (e.g., petroleum coke) with a combined mass of KOH and K₂CO₃ at a defined ratio (e.g., 1:1 strong:weak alkali).
- Single-Stage Activation Pyrolysis: Subject the mixture directly to pyrolysis in an inert atmosphere. The heating profile triggers the coordinated activation mechanism described in Section 3.1.
- Post-processing: Wash the product thoroughly with dilute HCl and deionized water to remove potassium salts and other impurities. Dry to obtain the final porous carbon.
Essential Materials and Reagents
Table 2: Research Reagent Solutions for Porous Carbon Synthesis
| Reagent / Material | Function in Synthesis | Exemplary Application |
|---|---|---|
| KOH (Potassium Hydroxide) | Strong alkali activator; creates high surface area and microporosity via corrosive etching [56] [57]. | Primary or co-activator in mixed alkali systems [56] [57]. |
| K₂CO₃ (Potassium Carbonate) | Weak alkali activator; modifies activation pathway for gentler, controlled micropore development [56]. | Co-activator with KOH to fine-tune microporosity and reduce overall corrosiveness [56]. |
| H₃PO₄ (Phosphoric Acid) | Chemical activator; promotes dehydration and cross-linking, often introducing oxygen functional groups [54]. | Activation of almond shells for hierarchical porous carbon [54]. |
| Biomass Precursors | Sustainable carbon source (e.g., almond shells, cedar wood, orange peel); inherent structure can template pores [54] [55] [57]. | Feedstock for eco-friendly and cost-effective porous carbon production. |
| Inert Gas (N₂ or Ar) | Creates an oxygen-free atmosphere during pyrolysis, preventing combustion and controlling reaction pathways. | Standard practice in all pyrolysis and activation protocols. |
Key Characterization Techniques
Rigorous characterization is essential to link synthesis parameters to material properties and performance.
- Surface Area and Porosity: Use N₂ adsorption-desorption isotherms analyzed with the Brunauer-Emmett-Teller (BET) method for specific surface area and Non-Local Density Functional Theory (NLDFT) for pore size distribution [54] [18].
- Structural Analysis: Raman Spectroscopy quantifies the degree of structural disorder (D band) versus graphitic order (G band). X-ray Diffraction (XRD) identifies crystalline phases and the amorphous nature of the carbon [55].
- Surface Chemistry: X-ray Photoelectron Spectroscopy (XPS) identifies and quantifies surface elemental composition and heteroatom functional groups (e.g., N, O) that enhance wettability and pseudocapacitance [18].
- Electrochemical Evaluation: Perform cyclic voltammetry (CV), galvanostatic charge-discharge (GCD), and electrochemical impedance spectroscopy (EIS) in two- or three-electrode cells to measure specific capacitance, rate capability, cycling stability, and ion transport resistance [54] [31].
Performance Data and Correlation with Synthesis Parameters
The ultimate validation of optimized synthesis lies in the electrochemical performance of the resulting porous carbon, particularly its ion adsorption capacity and energy storage capability.
Table 3: Correlation Between Synthesis Parameters and Electrochemical Performance
| Precursor (Activator) | Pyrolysis Temp. (°C) | Activator Ratio | Specific Surface Area (m²/g) | Specific Capacitance | Cycle Stability |
|---|---|---|---|---|---|
| Almond Shell (H₃PO₄) [54] | 1000 | H₃PO₄ : Biomass = 3:1 | 1164 - 1395 | 142 F/g (0.5 A/g) | 88.4% after 20,000 cycles |
| Petroleum Coke (KOH/K₂CO₃) [56] | Not Specified | KOH : K₂CO₃ = 1:1 | 1962 | 296.7 F/g (1 A/g) | 98.3% after 10,000 cycles |
| Coal Liquefaction Residue (KOH) [18] | Not Specified | KOH : (CDs+Melamine) = 1:2 | Data not provided | 314 F/g (1 A/g) | Data not provided |
| Cedar Wood (None) [55] | 900 | N/A | Data not provided | ~50% of Commercial AC | >95% after 10,000 cycles |
Optimizing pyrolysis temperature and activator ratios is a fundamental and impactful strategy for engineering porous carbon materials with enhanced ion adsorption and electrochemical performance. Key takeaways for researchers include:
- Pyrolysis Temperature must be optimized for the specific precursor-activator system, with a range of 900-1000°C often providing a favorable balance of conductivity, porosity, and surface functionality.
- Activator Selection and Ratio are powerful tools for fine-tuning pore architecture. Employing a mixed alkali system (KOH/K₂CO₃) can leverage synergistic effects, where KOH acts as a pore seed promoter and K₂CO₃ serves as a pathway modifier for controlled microporosity development.
- Synthesis Pathway choice between one-step and two-step activation involves a trade-off between performance and process complexity, which must be evaluated based on application requirements and sustainability goals.
The insights and experimental protocols compiled in this guide provide a foundation for the rational design of next-generation porous carbon electrodes. Future research will continue to refine these parameters and explore novel activator combinations to push the boundaries of energy storage and ion adsorption technology.
The Critical Role of Initial Solution pH on Ion Speciation and Adsorption Affinity
The efficacy of adsorption processes using porous materials is governed by a complex interplay of factors, among which the initial solution pH stands as a paramount and master variable. In the context of ion adsorption onto porous carbon electrodes, pH exerts a critical influence that extends from the macroscopic electrostatic environment to the molecular-scale speciation of ions and the chemistry of the adsorbent surface. This technical guide delves into the mechanistic role of pH, framing its impact within the broader research on advanced carbon electrodes for applications ranging from energy storage to environmental remediation. A profound understanding of these relationships is indispensable for the rational design of efficient adsorption systems.
The initial pH of a solution directly dictates the protonation state of oxygen-containing functional groups (e.g., carboxyl, carbonyl, hydroxyl) on the carbon surface, thereby controlling the magnitude and sign of the surface charge [58] [30]. This, in turn, establishes the electrostatic landscape that either attracts or repels target ions. Furthermore, pH determines the speciation of ions in solution, particularly for hydrolyzable metals, shifting the equilibrium between free aquated ions, hydrolyzed species, and precipitated hydroxides. The affinity of an ion for the carbon surface is thus not a fixed property but a function of the solution chemistry, which is dominantly controlled by pH.
Fundamental Mechanisms of pH Influence
The initial solution pH influences adsorption affinity through several interconnected mechanisms that collectively determine the outcome of the adsorption process.
Surface Charge and Point of Zero Charge (PZC)
The carbon-electrolyte interface acquires surface charge through two primary pathways: a reversible surface charge controlled by potential-determining ions (H⁺ and OH⁻), and a polarizable surface charge controlled by an externally applied potential (pE) [59]. The reversible charge is intrinsically linked to solution pH. The Point of Zero Charge (pH~PZC~) is the pH at which the carbon surface possesses a net neutral charge. At pH < pH~PZC~, the surface is protonated, acquiring a positive charge that favors anion adsorption. At pH > pH~PZC~, the surface deprotonates, becoming negatively charged and favoring cation adsorption [59] [60]. For instance, the YP-50F activated carbon exhibits a basic point of zero charge, indicating a negatively charged surface across a wide pH range, which enhances cation adsorption under neutral and basic conditions [30].
Ion Speciation and Functional Group Interactions
pH-induced changes in ion speciation directly alter adsorption affinity. For heavy metals like lead (Pb²⁺), adsorption capacity can significantly increase with pH, as observed with Manihot esculenta chaff, where higher pH reduces competition with H⁺ for surface sites and may promote hydrolysis of metal ions [60]. For carbon dioxide capture, the electrostatic attraction from specific oxygen-containing functional groups on the carbon surface is a key mechanism, with carboxyl groups identified as having the strongest attraction to CO₂ [58]. Solution pH during material synthesis can tailor these functional groups; highly acidic hydrothermal conditions (pH 1) yielded lignin-based porous carbon with the highest carboxyl group content and the superior CO₂ adsorption capacity of 5.10 mmol/g [58].
Table 1: Quantified Impact of pH on Adsorption Performance in Various Systems
| Adsorbent System | Target Ion/Molecule | pH Effect & Optimal Range | Quantified Impact | Primary Mechanism |
|---|---|---|---|---|
| Lignin-based Porous Carbon (LPC-pH1) [58] | CO₂ | Highest capacity at low pH (pH1) | 5.10 mmol/g at 0°C, 1 bar | Maximized carboxyl group content on carbon surface |
| Activated Carbon YP-50F [30] | H₃O⁺/TFSI⁻ | Uptake of H₃O⁺ and TFSI⁻ under acidic conditions (pH ~0.5) | Increased ion uptake & capacitance in acidic electrolytes | H₃O⁺ co-adsorption compensating for TFSI⁻ excess in pores |
| NSA@G Carbon Electrode [59] | Ca²⁺ | Reversible charge dominates at high pH; Polarizable charge at low pH & high pE | Reversible charge: 60-83% of uptake at high pH; Polarizable charge: 60-62% at low pH/high pE | pH-dependent reversible surface charge vs. potential-dependent polarizable charge |
| Manihot esculenta Chaff [60] | Pb²⁺ | Adsorption capacity increases with pH | Raw: 74.03 mg/g; Acid-modified: 96.28 mg/g | Reduced H⁺ competition, ion exchange, complexation with -OH, C=O |
Electrostatic and Non-Electrostatic Contributions
In electrochemical systems such as capacitive deionization (CDI), the interplay between pH-dependent (reversible) and potential-dependent (polarizable) charging is critical [59]. The contribution of each pathway to total ion uptake is not fixed but varies with the solution conditions. For Ca²⁺ electrosorption, the reversible surface charge governed by pH plays a more significant role at high pH values, contributing 60-83% of the total uptake. In contrast, the polarizable surface charge controlled by applied potential dominates at low pH and high potential, contributing 60-62% of the total Ca²⁺ uptake [59]. This quantification highlights that ignoring the pH-dependent reversible charge can lead to an incomplete and potentially inaccurate understanding of electrosorption mechanisms.
Experimental Protocols for Investigating pH Effects
A robust experimental framework is essential to isolate and understand the role of initial solution pH. The following protocols outline key methodologies.
Determining the Point of Zero Charge (PZC)
The pH~PZC~ is a fundamental property of an adsorbent material.
- Procedure: Prepare a series of 0.01 M NaCl solutions (or an inert electrolyte of choice). Adjust the initial pH (pHi) of each solution over a wide range (e.g., 2-12) using small volumes of HCl or NaOH. Add a fixed mass of the adsorbent (e.g., 35 mg) to each solution and agitate the mixtures for a sufficient time to reach equilibrium (e.g., 3 h) [60]. Measure the final pH (pHf) of each solution.
- Analysis: Plot the difference between the initial and final pH (ΔpH = pHf - pHi) against the initial pH (pHi). The point at which the curve crosses ΔpH = 0 is the PZC. At this pH, the surface does not significantly alter the solution pH.
Batch Adsorption Experiments as a Function of pH
This protocol evaluates the adsorption affinity and capacity across a pH range.
- Procedure: Prepare a series of solutions containing a fixed, known initial concentration of the target ion. Adjust each solution to a different initial pH covering the range of interest. Introduce a constant mass of the adsorbent into each solution and agitate in a controlled environment (constant temperature, shaking speed) until equilibrium is reached. Sample the supernatant, separate it from the adsorbent, and analyze the equilibrium concentration of the ion using appropriate techniques (e.g., AAS, ICP-OES, ion chromatography).
- Analysis: The amount adsorbed at equilibrium, q~e~ (mg/g), is calculated. Plotting q~e~ versus the initial or equilibrium pH reveals the pH profile for optimal adsorption. This directly links pH to adsorption performance.
Probing Ion Uptake and Speciation with Solid-State NMR
Nuclear Magnetic Resonance (NMR) spectroscopy can provide a molecular-level picture of ion adsorption in porous carbons, even quantifying the uptake of challenging species like H₃O⁺ [30].
- Procedure: The carbon material is equilibrated with an aqueous electrolyte (e.g., LiTFSI) at a specific pH. The sample is then packed into an NMR rotor. ¹H, ⁷Li, or ¹⁹F NMR spectra are acquired.
- Analysis: Resonances from species adsorbed inside the carbon pores are distinguished from those in the bulk electrolyte by their chemical shift. Deconvolution of the spectra based on a two-site exchange model allows for accurate quantification of the adsorbed ions (Li⁺, TFSI⁻, H₃O⁺). This methodology was pivotal in demonstrating H₃O⁺ co-adsorption in carbon pores under acidic conditions, which is necessary for charge balance when anion adsorption exceeds cation adsorption [30].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 2: Key Reagents and Materials for Ion Adsorption Studies
| Reagent/Material | Function & Rationale | Example from Literature |
|---|---|---|
| Sodium Lignosulfonate | Sustainable, renewable carbon precursor for synthesizing lignin-based porous carbons (LPCs). | Used to prepare LPCs for CO₂ adsorption studies [58]. |
| Strong Acids/Bases (H₂SO₄, HNO₃, NaOH) | Adjust initial solution pH for adsorption experiments; chemically modify adsorbent surfaces (e.g., acid modification introduces oxygen-containing groups). | HNO₃ used to modify cassava chaff, enhancing Pb²⁺ capacity [60]; H₂SO₄ for hydrothermal pH adjustment [58]. |
| Inert Electrolyte (e.g., NaCl, NaNO₃) | Maintains constant ionic strength during pH/PZC experiments, ensuring changes are due to H⁺/OH⁻ and not ionic strength variation. | 0.01 M NaCl used for PZC determination of cassava chaff [60]. |
| Model Aqueous Pollutants (e.g., Pb²⁺, Ca²⁺ salts) | Representative target ions for evaluating adsorption performance, kinetics, and isotherms under controlled pH. | Ca(ClO₄)₂ for alkaline earth metal electrosorption [59]; Lead salts for heavy metal adsorption [60]. |
| Commercial Activated Carbons (e.g., YP-50F, BAU-A) | Well-characterized, standard porous carbon materials used as benchmarks or precursors for functionalized adsorbents. | YP-50F used for NMR studies of H₃O⁺ uptake [30]; BAU-A as a precursor for magnetic adsorbents [61]. |
Visualization of pH-Dependent Adsorption Mechanisms
The following diagram synthesizes the core concepts and experimental pathways for investigating the role of initial solution pH.
This diagram illustrates how the initial solution pH acts as a central control point, simultaneously influencing the adsorbent's surface charge, the chemical state of the target ions, and the protonation of surface functional groups. These factors converge to determine the net adsorption affinity. The experimental pathway (bottom) provides a systematic approach to unravel these complex interactions and arrive at a molecular-level understanding of the governing mechanisms.
The initial solution pH is a powerful, non-invasive parameter that critically controls the efficiency and mechanism of ion adsorption onto porous carbon materials. Its influence permeates every aspect of the adsorption process, from dictating the electrostatic potential at the carbon-solution interface to determining the speciation of ions in solution and the availability of specific binding sites. A deliberate and systematic investigation of pH effects—through PZC determination, batch adsorption studies, and advanced characterization techniques like NMR—is not merely a procedural step but a fundamental requirement. For researchers and scientists designing next-generation porous carbon electrodes, mastering the critical role of initial solution pH is the key to unlocking superior control over ion selectivity, adsorption capacity, and overall system performance.
Understanding the kinetics of ion adsorption is fundamental to optimizing the performance of porous carbon electrodes in applications such as capacitive deionization (CDI), a promising energy-efficient desalination technology [9]. Kinetic analysis provides critical insights into the rate of solute uptake, the time required to reach equilibrium, and the underlying mechanisms controlling the adsorption process. For researchers developing advanced porous carbon materials, applying kinetic models is indispensable for evaluating and designing efficient systems. The pseudo-first-order (PFO) and pseudo-second-order (PSO) models are among the most prevalent for analyzing experimental data, offering a mathematical framework to quantify adsorption rates and propose potential mechanisms [62]. This guide details the theoretical foundation, experimental protocols, and data interpretation for applying these models within the context of ion adsorption on porous carbons.
Theoretical Foundations of Kinetic Models
The Pseudo-First-Order (PFO) Model
- Conceptual Basis: The PFO model, originally developed by Lagergren, assumes that the rate of adsorption is proportional to the difference between the equilibrium adsorption capacity and the adsorbed amount at any time, ( t ) [62]. The prefix "pseudo" indicates that the model is applied to solid-liquid adsorption systems, even though its form is analogous to a first-order chemical reaction.
- Differential Form: ( \frac{dqt}{dt} = k1 (qe - qt) )
- Integrated Form: ( \log(qe - qt) = \log(qe) - \frac{k1}{2.303}t )
- Application: A linear plot of ( \log(qe - qt) ) versus ( t ) suggests the adsorption process follows PFO kinetics. The slope and intercept are used to determine the rate constant, ( k1 ), and the theoretical equilibrium capacity, ( qe ).
The Pseudo-Second-Order (PSO) Model
- Conceptual Basis: The PSO model assumes that the adsorption rate is proportional to the square of the number of unoccupied sites [62]. A literal interpretation of its mechanism suggests the rate-limiting step may involve the "collision" of two independent unoccupied sites on the adsorbent surface.
- Differential Form: ( \frac{dqt}{dt} = k2 (qe - qt)^2 )
- Integrated Form: ( \frac{t}{qt} = \frac{1}{k2 qe^2} + \frac{1}{qe}t )
- Application: The PSO model is widely applied due to its excellent fit to diverse experimental adsorption data [62]. A linear plot of ( t/qt ) versus ( t ) indicates conformity with the model, allowing ( qe ) and ( k_2 ) to be calculated from the slope and intercept.
Model Selection and Mechanistic Interpretation
While the PSO model often provides a superior fit to experimental data for cellulosic and carbon-based adsorbents [62], a good statistical fit does not automatically validate its underlying mechanistic assumption. The adsorption kinetics on porous materials is often governed by diffusion-limited processes, influenced by heterogeneous pore size distributions and the partitioning of solute between dissolved and adsorbed states [62]. Therefore, kinetic model fitting should be complemented with diffusion model analysis (e.g., intraparticle diffusion) to gain a more comprehensive mechanistic understanding [63]. The fractional-order Avrami's model is another alternative that can provide a more accurate description of complex processes involving multiple pathways [63].
Table 1: Key Parameters of Pseudo-First-Order and Pseudo-Second-Order Kinetic Models
| Parameter | Pseudo-First-Order (PFO) | Pseudo-Second-Order (PSO) |
|---|---|---|
| Rate Law | ( \frac{dqt}{dt} = k1 (qe - qt) ) | ( \frac{dqt}{dt} = k2 (qe - qt)^2 ) |
| Integrated Form | ( \log(qe - qt) = \log(qe) - \frac{k1}{2.303}t ) | ( \frac{t}{qt} = \frac{1}{k2 qe^2} + \frac{1}{qe}t ) |
| Linear Plot | ( \log(qe - qt) ) vs. ( t ) | ( t/q_t ) vs. ( t ) |
| Rate Constant | ( k_1 ) (min⁻¹) | ( k_2 ) (g/mg·min) |
| Equilibrium Capacity | ( q_e ) (from intercept) | ( q_e ) (from slope) |
| Primary Assumption | Rate is driven by site availability | Rate is driven by square of unoccupied sites |
Experimental Protocols for Kinetic Studies
Workflow for Adsorption Kinetics
The following diagram outlines the standard experimental workflow for conducting kinetic analysis of ion adsorption onto porous carbon electrodes.
Detailed Methodologies for Key Experiments
Batch Adsorption Experiment
This is the core experiment for generating kinetic data.
- Procedure:
- Adsorbent Preparation: Weigh a precise mass (e.g., 10-50 mg) of the porous carbon electrode material. The material should be pre-dried and characterized for properties like specific surface area (SBET) and pore size distribution (PSD), as PSD is a critical parameter determining adsorption capacity and rate [63].
- Solution Preparation: Prepare a known volume (e.g., 100-500 mL) of the adsorbate solution (e.g., NaCl for CDI studies) at a predetermined initial concentration (C₀) in a temperature-controlled environment.
- Initiating Adsorption: Add the weighed adsorbent to the solution under constant agitation to ensure uniform mixing and eliminate external mass transfer limitations.
- Sampling: At predetermined time intervals (e.g., 1, 2, 5, 10, 20, 30, 60 min), withdraw small aliquots of the solution.
- Separation: Immediately separate the adsorbent from the liquid aliquot using filtration (e.g., 0.45 μm syringe filter) or centrifugation.
- Analysis: Measure the concentration of the adsorbate in the filtrate (Cₜ) using an appropriate analytical technique (e.g., conductivity meter for salts, UV-Vis spectrophotometry for dyes, Atomic Absorption Spectroscopy for metal ions).
- Data Calculation: The amount of adsorbate adsorbed per unit mass of adsorbent at time ( t ), ( qt ) (mg/g), is calculated as: ( qt = \frac{(C0 - Ct) \times V}{m} ), where ( V ) is the volume of the solution (L), and ( m ) is the mass of the adsorbent (g).
Data Fitting and Model Validation
- Procedure:
- Plot Experimental Data: Plot the calculated ( qt ) values against time ( t ) to visualize the adsorption profile.
- Linear Regression: For the PFO model, plot ( \log(qe - qt) ) versus ( t ). For the PSO model, plot ( t/qt ) versus ( t ). The experimental ( qe ) value is required for the PFO plot, whereas the PSO model calculates ( qe ) internally.
- Evaluate Linearity: Perform linear regression on both plots. The model with a correlation coefficient (R²) closer to 1.0 and whose predicted ( q{e,calc} ) value more closely matches the experimental ( q{e,exp} ) is generally considered the better fit.
- Parameter Extraction: Calculate the kinetic parameters ( k1 ) or ( k2 ) and ( q_{e,calc} ) from the slope and intercept of the linear plots.
Table 2: Example Kinetic Data for CO₂ Adsorption on Activated Hydrochars (at 25 °C) [63]
| Adsorbent | Experimental qₑ (mg/g) | PFO k₁ (min⁻¹) | PFO R² | PSO k₂ (g/mg·min) | PSO R² | Avrami Model R² |
|---|---|---|---|---|---|---|
| Hydrochar (KHCO₃) | 145.2 | - | - | - | - | Best Fit [63] |
| Hydrochar (Physical) | ~110 (approx.) | - | - | - | - | Best Fit [63] |
| Commercial AC | ~90 (approx.) | - | - | - | - | Best Fit [63] |
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Reagents and Materials for Adsorption Kinetic Studies
| Item | Function & Importance | Examples & Notes |
|---|---|---|
| Porous Carbon Material | The core adsorbent; its properties dictate performance. | Activated hydrochars [63], hierarchical porous carbons (HPCs) [9]. Key properties: SBET, pore volume, surface chemistry. |
| Activating Agents | Chemicals used to create and tune the porous structure. | KHCO₃, KOH, H₃PO₄, ZnCl₂. Trend toward greener activators (e.g., KHCO₃) to reduce environmental impact and equipment corrosion [63] [9]. |
| Target Adsorbate | The ion or molecule to be removed from solution. | NaCl (for CDI), dyes (e.g., methylene blue), heavy metal ions (e.g., Cu²⁺, Pb²⁺). |
| Analytical Instrumentation | To quantify adsorbate concentration over time. | Conductivity meter, UV-Vis Spectrophotometer, Inductively Coupled Plasma (ICP) instruments. |
| Agitation System | To ensure proper mixing and minimize external diffusion. | Mechanical shaker, magnetic stirrer with temperature control (e.g., water bath). |
| Filtration/Separation | To separate adsorbent from solution at timed intervals. | Syringe filters (0.45 μm), centrifuge. Critical for accurate Cₜ measurement. |
The application of PFO and PSO kinetic models is a critical step in characterizing the adsorption performance of porous carbon electrodes. While the PSO model frequently demonstrates an excellent fit for adsorption data on carbon and cellulosic materials, researchers must be cautious in interpreting this as direct proof of a chemical reaction-based mechanism. The kinetics are often dominated by diffusion processes through a heterogeneous pore network [62]. A robust analysis involves fitting multiple models, validating predicted equilibrium capacities against experimental data, and employing complementary techniques like intraparticle diffusion modeling [63] to unravel the true rate-controlling steps. This comprehensive approach provides the insights necessary to engineer next-generation porous carbon materials with tailored kinetics for enhanced performance in water treatment, energy storage, and related applications.
Adsorption isotherms are fundamental mathematical models that describe the equilibrium distribution of adsorbate ions between a liquid phase and a solid adsorbent surface at a constant temperature. For researchers developing advanced porous carbon electrodes, these models provide critical insights into the adsorption capacity, affinity, and underlying mechanisms that govern contaminant removal and ion selectivity in water treatment and resource recovery applications. The modeling of adsorption equilibrium is particularly crucial in the context of capacitive deionization (CDI), a promising brackish water desalination technology where ions are electrostatically removed and stored in the electrical double layers of porous carbon electrodes [64]. Within this technological framework, the Langmuir and Freundlich isotherms emerge as two of the most extensively applied models for interpreting experimental data, each founded on distinct physical assumptions about the adsorbent surface and the adsorption process.
The selection of an appropriate isotherm model is not merely a statistical exercise; it provides fundamental insights into the adsorption mechanisms and the energetic heterogeneity of the adsorbent surface. This interpretation directly informs material design choices, enabling researchers to tailor carbon electrodes with specific surface properties, pore architectures, and surface chemistries to enhance performance for targeted applications. For instance, the presence of specific functional groups on carbon surfaces can significantly influence metal ion selectivity, as demonstrated in studies of superparamagnetic alginate beads containing ordered mesoporous carbon (CMK-3), where the presence of O=C bonds and other elements like F− contributed to superior adsorption efficiency for Cd(II), Hg(II), and Ni(II) ions [65]. This technical guide provides an in-depth comparison of the Langmuir and Freundlich models, detailing their theoretical foundations, mathematical formulations, experimental application protocols, and interpretation within the specific context of ion adsorption on porous carbon electrodes.
Theoretical Foundations and Mathematical Formulations
The Langmuir Adsorption Model
The Langmuir isotherm, developed by Irving Langmuir, represents a fundamental theoretical model based on a kinetic perspective and the principle of adsorption dynamic equilibrium, where the rate of adsorption equals the rate of desorption [66]. This model operates on several key assumptions: (1) adsorption occurs in a monomolecular layer (monolayer coverage); (2) the adsorbent surface is homogeneous, meaning all adsorption sites are energetically identical; (3) there is no interaction between adsorbed molecules on adjacent sites; and (4) the adsorption energy is constant across all sites [66] [67]. The model is particularly applicable to chemisorption processes where strong chemical bonds form between the adsorbate and adsorbent [66].
The non-linear form of the Langmuir isotherm equation is represented as:
[ qe = \frac{q{\text{max}} KL Ce}{1 + KL Ce} ]
where:
- ( q_e ) is the amount of solute adsorbed per unit weight of adsorbent at equilibrium (mg/g)
- ( q_{\text{max}} ) is the maximum monolayer adsorption capacity (mg/g)
- ( K_L ) is the Langmuir adsorption constant related to adsorption energy (L/mg)
- ( C_e ) is the equilibrium concentration of the solute in the bulk solution (mg/L) [66]
For linearization and parameter determination, the equation can be rearranged into several linear forms. The most common linear form is:
[ \frac{Ce}{qe} = \frac{1}{KL q{\text{max}}} + \frac{Ce}{q{\text{max}}} ]
A plot of ( Ce/qe ) versus ( Ce ) yields a straight line with a slope of ( 1/q{\text{max}} ) and an intercept of ( 1/(KL q{\text{max}}) ), from which the parameters ( q{\text{max}} ) and ( KL ) can be calculated [66]. The Langmuir model also incorporates a dimensionless equilibrium parameter, ( R_L ), defined as:
[ RL = \frac{1}{1 + KL C_0} ]
where ( C0 ) is the initial adsorbate concentration. The ( RL ) value indicates the nature of the adsorption process: unfavorable (( RL > 1 )), linear (( RL = 1 )), favorable (( 0 < RL < 1 )), or irreversible (( RL = 0 )) [67].
For complex systems such as natural soils or engineered carbon surfaces that exhibit multiple binding site energies, the basic Langmuir model has been extended to a two-surface equation:
[ qe = \frac{K{L1} q{m1} Ce}{1 + K{L1} Ce} + \frac{K{L2} q{m2} Ce}{1 + K{L2} C_e} ]
where ( q{m1} ) and ( K{L1} ) represent the adsorption maximum and equilibrium constant for low-energy surfaces, and ( q{m2} ) and ( K{L2} ) correspond to high-energy surfaces [66]. The total adsorption maximum becomes ( qm = q{m1} + q_{m2} ). This refinement is particularly relevant for heterogeneous porous carbon electrodes where different surface functional groups and pore structures create distinct adsorption environments with varying energies.
The Freundlich Adsorption Model
In contrast to the Langmuir model, the Freundlich isotherm is an empirical model developed to describe adsorption on heterogeneous surfaces with non-uniform distribution of adsorption heat [67]. It does not assume monolayer coverage and is more applicable to physical adsorption processes (physisorption) and multi-layer adsorption. The model is particularly useful for representing adsorption on complex natural adsorbents and engineered materials with surface irregularities, such as activated carbons and functionalized porous carbon electrodes.
The non-linear form of the Freundlich equation is expressed as:
[ qe = KF C_e^{1/n} ]
where:
- ( q_e ) is the amount of solute adsorbed per unit weight of adsorbent at equilibrium (mg/g)
- ( K_F ) is the Freundlich constant representing adsorption capacity ((mg/g)(L/mg)¹/ⁿ)
- ( 1/n ) is the heterogeneity factor indicative of adsorption intensity
- ( C_e ) is the equilibrium concentration of the solute (mg/L) [67] [68]
The linearized form of the equation is:
[ \log qe = \log KF + \frac{1}{n} \log C_e ]
A plot of ( \log qe ) versus ( \log Ce ) yields a straight line with a slope of ( 1/n ) and an intercept of ( \log K_F ). The value of ( 1/n ) provides information about the favorability and nature of the adsorption process. For favorable adsorption, ( 0 < 1/n < 1 ), while ( 1/n > 1 ) indicates unfavorable adsorption, and ( 1/n = 1 ) represents linear adsorption [67]. If ( 1/n = 0 ), the adsorption process is considered irreversible.
Unlike the Langmuir model, the Freundlich equation does not predict saturation of the adsorbent surface, meaning it lacks a theoretical maximum adsorption capacity. However, Zeldowitch's theoretical work demonstrated that the Freundlich model can be extended to describe saturation behavior by considering the distribution of adsorption sites with different energies [68]. According to this theory, at high concentrations (( x \gg b_0 )), the coverage approaches saturation in a Langmuir-like form:
[ \frac{q(x)}{q{\infty}} \approx \frac{x}{x + b0/(1 + 1/\alpha)} ]
where ( b_0 ) is a constant related to the adsorption energy distribution [68]. This extension bridges the gap between the power-law behavior at low concentrations and the saturation regime at high concentrations, providing a more comprehensive framework for analyzing experimental data across the entire concentration range.
Comparative Analysis of Model Characteristics
Table 1: Fundamental Characteristics of Langmuir and Freundlich Isotherm Models
| Parameter | Langmuir Model | Freundlich Model |
|---|---|---|
| Theoretical Basis | Theoretical (kinetic and thermodynamic principles) | Empirical (experimental data fitting) |
| Surface Assumption | Homogeneous surface with identical sites | Heterogeneous surface with different site energies |
| Adsorption Layer | Monolayer coverage | Multilayer capability |
| Adsorption Mechanism | Primarily chemisorption | Primarily physisorption |
| Interaction Between Adsorbed Molecules | No interaction assumed | Accounts for adsorbate-adsorbate interactions |
| Saturation Behavior | Predicts clear saturation plateau ((q_{\text{max}})) | No saturation limit in original form |
| Temperature Dependence | Constants change predictably with temperature | Constants change empirically with temperature |
| Mathematical Form | ( qe = \frac{q{\text{max}} KL Ce}{1 + KL Ce} ) | ( qe = KF C_e^{1/n} ) |
| Linear Form | ( \frac{Ce}{qe} = \frac{1}{KL q{\text{max}}} + \frac{Ce}{q{\text{max}}} ) | ( \log qe = \log KF + \frac{1}{n} \log C_e ) |
Table 2: Practical Application Guidelines for Model Selection
| Criterion | Langmuir Model Preferred When | Freundlich Model Preferred When |
|---|---|---|
| Surface Character | Homogeneous surfaces (e.g., well-defined carbon materials) | Heterogeneous surfaces (e.g., activated carbon, functionalized materials) |
| Adsorbate Concentration | Moderate to high concentrations approaching saturation | Low to moderate concentrations |
| System Complexity | Single-component systems with specific binding | Multi-component systems with competitive adsorption |
| Data Pattern | Data shows clear plateau at high concentrations | Data follows power-law relationship without clear plateau |
| Application Examples | Cd(II), Ni(II) adsorption on CMK-3 carbon [65]; RO16 dye on Cu(I)-PANI composite [69] | Hg(II) adsorption on activated charcoal cloth [64]; NaCl on carbon nanotubes at low concentrations [64] |
The fundamental distinction between the Langmuir and Freundlich models lies in their conceptualization of the adsorbent surface. The Langmuir model envisions a homogeneous surface where all sites possess equal energy, leading to identical adsorption affinities across the entire surface. This assumption is more valid for highly engineered carbon materials with uniform surface chemistry, such as the ordered mesoporous CMK-3 carbon used in superparamagnetic beads for heavy metal removal [65]. In contrast, the Freundlich model accommodates surface heterogeneity, recognizing that real adsorbent surfaces typically contain sites with different energies, which is particularly true for activated carbons with complex pore structures and varied surface functional groups.
Regarding adsorption capacity prediction, the Langmuir model provides a definitive maximum adsorption capacity ((q{\text{max}})), representing the point at which a complete monolayer forms and no further adsorption can occur. This parameter is highly valuable for comparing different adsorbents and designing treatment systems with predictable capacity limits. For instance, in a study on Cu(I)-polyaniline composite for RO16 dye removal, the Langmuir model yielded a (q{\text{max}}) value of 392.156 mg/g, providing a clear benchmark for performance evaluation [69]. The Freundlich model, being empirical, lacks this theoretical maximum but effectively describes adsorption across a wide concentration range, particularly at lower concentrations where surface heterogeneity significantly influences adsorption behavior.
The nature of the adsorption process also guides model selection. The Langmuir model is more appropriate for chemisorption processes involving strong, specific chemical interactions, such as the surface complexation mechanisms observed in heavy metal adsorption on functionalized carbon surfaces [65] [66]. The Freundlich model better describes physisorption processes dominated by weaker, non-specific interactions like van der Waals forces, which are common in organic contaminant adsorption on activated carbons.
Experimental Protocols for Isotherm Determination
Materials Preparation and Characterization
The foundation of reliable adsorption isotherm data begins with meticulous preparation and characterization of adsorbent materials. For research on porous carbon electrodes, this typically involves synthesis or procurement of carbon materials with controlled properties. The synthesis of composite adsorbents often follows established protocols, such as the in-situ polymerization and composite formation (IPCF) technique used for creating Cu(I)-polyaniline composites, where aniline solution is mixed with copper sulfate in methanol and stirred continuously for 24 hours to form parrot-green precipitates [69]. Similarly, superparamagnetic alginate beads containing ordered mesoporous carbon (CMK-3) or commercial activated carbon are prepared through specific encapsulation methods to create magnetically recoverable adsorbents [65].
Comprehensive characterization of the adsorbent materials is essential for correlating physical and chemical properties with adsorption performance. Key characterization techniques include:
- Surface area and porosity analysis using Brunauer-Emmett-Teller (BET) method with instruments like mercury intrusion porosimetry (e.g., AutoPore IV 9500) to determine specific surface area, pore volume, and pore size distribution [69]
- Structural analysis through powder X-ray diffraction (XRD) using instruments like XRD 6000 to confirm crystallinity and phase composition [69]
- Surface chemistry analysis via X-ray photoelectron spectroscopy (XPS) to identify elemental composition and functional groups (e.g., O=C bonds, F− content) that influence adsorption behavior [65] [69]
- Thermal stability assessment using thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) to understand material stability under operational conditions [69]
Table 3: Essential Research Reagents and Materials for Adsorption Studies
| Material/Reagent | Specification | Function in Research |
|---|---|---|
| Porous Carbon Materials | Ordered mesoporous carbon (CMK-3), Activated Carbon | Primary adsorbent providing surface area for ion adsorption |
| Polymer Matrix | Sodium Alginate, Polyaniline (PANI) | Structural component for bead formation or composite enhancement |
| Cross-linking Agents | Calcium chloride, Copper sulfate | Facilitates formation of stable adsorbent structures |
| Target Adsorbates | Cd(II), Ni(II), Hg(II) salts; RO16 dye | Model contaminants for evaluating adsorption performance |
| Solvents | Deionized water, Methanol | Medium for synthesis and adsorption experiments |
| pH Adjusters | HCl, NaOH solutions | Control solution pH to study its effect on adsorption |
| Analysis Instruments | UV-Vis Spectrophotometer, AAS, ICP-MS | Quantify adsorbate concentration before and after adsorption |
Batch Adsorption Experiments
The core experimental procedure for generating adsorption isotherm data involves batch equilibrium studies. A typical protocol follows these steps:
Stock Solution Preparation: Dissolve the target adsorbate (e.g., heavy metal salts or dyes) in deionized water to create a concentrated stock solution (e.g., 500 mg/L for RO16 dye) [69].
Calibration Curve Development: Prepare a series of standard solutions with known concentrations (e.g., 20, 40, 60, 80, and 100 mg/L) and measure their absorbance using UV-Vis spectroscopy or other appropriate analytical techniques to establish a relationship between concentration and instrumental response [69].
Experimental Series Setup: Prepare a series of Erlenmeyer flasks or centrifuge tubes containing fixed amounts of adsorbent (e.g., 0.1 g) and varying initial concentrations of the adsorbate solution, keeping the solution volume constant across all samples. The concentration range should be designed to cover from minimal adsorption to near-saturation conditions.
Equilibrium Process: Agitate the mixtures in a temperature-controlled shaker at constant speed (e.g., 150 rpm) for a predetermined period sufficient to reach equilibrium, typically 24 hours based on kinetic studies [69].
Phase Separation: After the equilibrium period, separate the solid adsorbent from the liquid phase using centrifugation or filtration. For magnetic adsorbents like superparamagnetic alginate beads, external magnetic fields can be employed for efficient recovery [65].
Residual Concentration Analysis: Measure the equilibrium concentration of the adsorbate in the supernatant using appropriate analytical techniques (UV-Vis spectroscopy for dyes, atomic absorption spectroscopy (AAS) or inductively coupled plasma mass spectrometry (ICP-MS) for metals).
Data Calculation: Calculate the amount adsorbed at equilibrium ((q_e)) using the mass balance equation:
[ qe = \frac{(C0 - C_e) V}{m} ]
where:
- ( C_0 ) = initial concentration (mg/L)
- ( C_e ) = equilibrium concentration (mg/L)
- ( V ) = solution volume (L)
- ( m ) = mass of adsorbent (g)
Data Fitting and Model Validation
Once experimental data is collected, the process of model fitting and validation begins:
Linearization and Plotting: For initial assessment, plot the data according to both Langmuir ((Ce/qe) vs. (Ce)) and Freundlich ((\log qe) vs. (\log C_e)) linear forms.
Parameter Calculation: Determine model parameters from the slopes and intercepts of the linear plots. For Langmuir: (q{\text{max}} = 1/\text{slope}) and (KL = \text{intercept} \times \text{slope}). For Freundlich: (1/n = \text{slope}) and (K_F = 10^{\text{intercept}}).
Non-linear Regression: For more accurate parameter estimation, use non-linear regression analysis to fit the experimental data directly to the non-linear forms of the isotherm equations, as this avoids potential distortions introduced by linearization.
Goodness-of-Fit Evaluation: Assess the quality of fit using statistical parameters such as correlation coefficient (R²), residual sum of squares (RSS), and error functions like chi-square (χ²) test. As highlighted in research on magnetic hybrid beads, optimization analysis of isotherm models using both linearization and non-linear approaches with error functions provides more reliable parameter estimation [65].
Model Validation: Validate the selected model by comparing predicted values with experimental data not used in the parameter estimation process, or through cross-validation techniques.
Data Interpretation in Context of Porous Carbon Electrodes
Relating Isotherm Parameters to Material Properties
The parameters derived from adsorption isotherms provide valuable insights into the relationship between carbon electrode properties and adsorption performance. The Langmuir (q_{\text{max}}) parameter represents the theoretical monolayer capacity, which is directly influenced by the specific surface area and accessible pore volume of the carbon material. In capacitive deionization applications, this translates to the salt storage capacity of electrodes [64]. Research has shown that carbon materials with ordered mesoporous structures like CMK-3 often exhibit superior adsorption capacities compared to conventional activated carbons due to their enhanced accessibility to adsorption sites [65].
The Langmuir (K_L) constant reflects the affinity between the adsorbate and adsorbent surface, with higher values indicating stronger interactions. This parameter is particularly important in systems where selective ion removal is desired, such as in the recovery of valuable metals or the targeted removal of specific contaminants. Studies on superparamagnetic beads have demonstrated that materials containing CMK-3 carbon showed higher affinity for Cd(II) and Ni(II) ions, while those with commercial activated carbon exhibited greater affinity for Hg(II) ions, highlighting the role of surface chemistry in ion selectivity [65].
In the Freundlich model, the (K_F) parameter serves as an indicator of adsorption capacity, while the (1/n) parameter reflects the surface heterogeneity and adsorption intensity. Values of (1/n) less than 1 indicate favorable adsorption conditions, while values approaching zero suggest high surface heterogeneity. For porous carbon electrodes, the heterogeneity factor often correlates with the diversity of surface functional groups and pore size distributions. Research has shown that at low salt concentrations, the electrosorption on carbon nanomaterials may follow the Freundlich isotherm, as the adsorbed layer does not fulfill the monolayer capacity in diluted solutions [64].
Understanding Adsorption Mechanisms
Isotherm modeling provides crucial evidence for elucidating the fundamental mechanisms governing ion adsorption on carbon electrodes. The better fit of experimental data to a particular model offers insights into whether the adsorption occurs primarily through chemical-specific interactions (Langmuir-type) or physical-nonspecific interactions (Freundlich-type). However, real systems often involve multiple simultaneous mechanisms, as demonstrated in studies of heavy metal adsorption on magnetic hybrid beads, where adsorption occurred through "several mechanisms involving surface complexation, ion-exchange, precipitation, physical and chemical processes" [65].
The extension of Freundlich analysis to saturation regimes using Zeldowitch's theory enables researchers to bridge the gap between low-coverage power-law behavior and high-coverage saturation, providing a more comprehensive understanding of adsorption across the entire concentration range [68]. This approach is particularly valuable for designing carbon electrodes that operate effectively across varying concentration conditions, from trace contaminant removal to high-salinity environments.
In the context of capacitive deionization, where electrical fields enhance ion adsorption, the interpretation of isotherm parameters must consider the additional driving force. Studies have shown that the Langmuir isotherm often describes electrosorption data well, suggesting that monolayer adsorption assumptions may be valid for these systems [64]. However, the interplay between electrical double layer formation and specific chemical interactions creates complex adsorption environments that may require modified or hybrid isotherm models for accurate representation.
The selection between Langmuir and Freundlich isotherm models for understanding adsorption equilibrium on porous carbon electrodes depends fundamentally on the surface characteristics of the adsorbent and the nature of the adsorbate-adsorbent interactions. The Langmuir model provides a theoretically grounded framework for describing monolayer adsorption on homogeneous surfaces, offering clearly interpretable parameters for maximum capacity ((q{\text{max}})) and affinity ((KL)) that are invaluable for comparing materials and predicting system performance under saturation conditions. Conversely, the Freundlich model offers empirical flexibility for representing adsorption on heterogeneous surfaces, with parameters that effectively capture the complexity of real carbon materials with diverse surface sites and multilayer adsorption capabilities.
For researchers developing advanced porous carbon electrodes for environmental applications and capacitive deionization, a strategic approach involving both models, complemented by advanced characterization techniques, provides the most comprehensive understanding of adsorption behavior. The integration of Zeldowitch's theoretical extension to the Freundlich model further enhances its utility by bridging low-concentration power-law behavior with high-concentration saturation regimes. As carbon materials continue to evolve in complexity and functionality, with engineered surface chemistries and hierarchical pore structures, the sophisticated application of these fundamental isotherm models remains essential for rational material design and optimization of adsorption processes for specific ion separation challenges.
The performance of electrochemical devices for energy storage and environmental remediation, such as supercapacitors and capacitive deionization (CDI) systems, is fundamentally governed by the properties of their electrode materials. Porous carbon electrodes, in particular, have garnered significant attention due to their high specific surface area, excellent electrical conductivity, and structural versatility. However, their widespread application is hindered by three interconnected challenges: insufficient surface activity, limited hydrophilicity, and low ion selectivity. These limitations become especially critical in complex ionic environments, such as those encountered in the selective removal of heavy metals from wastewater or the recovery of valuable resources. Insufficient surface activity results in low charge storage capacity and sluggish charge transfer. Poor hydrophilicity impedes electrolyte wetting, limiting access to the extensive internal surface area of porous carbons. Perhaps most critically, a lack of ion selectivity prevents targeted removal of specific ions from multi-component solutions, a necessity for both environmental remediation and resource recovery applications. This whitepaper synthesizes recent scientific advances to provide a technical guide on overcoming these challenges through strategic material design, functionalization, and precise characterization. The insights are framed within the context of a broader research thesis on ion adsorption in porous carbon electrodes, aiming to equip researchers with the methodologies to develop next-generation electrochemical systems.
Material Design and Functionalization Strategies
Enhancing the performance of porous carbon electrodes requires a multi-faceted approach that targets their chemical, physical, and electronic properties. The following strategies have proven effective in addressing the core challenges.
Surface Functionalization with Polymers and Functional Groups
Modifying the carbon surface with specific polymers or functional groups is a direct method to enhance hydrophilicity and introduce selective binding sites.
- Cation-Exchange Polymer Composites: The fabrication of a composite film using two sulfonate-rich polymers, Nafion and poly(sodium 4-styrenesulfonate) (PSS), on a screen-printed electrode (SPE) demonstrates a synergistic approach [70]. The hydrophilic PSS component significantly improves surface wettability, facilitating better interaction with aqueous solutions and enhancing the mass transport of target metal ions. Concurrently, Nafion provides a selective cation-exchange function, preferentially allowing cations like Pb²⁺ and Cd²⁺ to reach the electrode surface while excluding anionic interferents [70]. This combination resulted in detection limits as low as 6.478 ppb for Pb²⁺ and 5.277 ppb for Cd²⁺.
- Heteroatom Doping: Introducing heteroatoms such as nitrogen (N) and oxygen (O) into the carbon lattice is a powerful strategy to modulate surface chemistry and electronic structure. Nitrogen/oxygen-codoped dense porous carbons (NDPCs) have been developed, where the N/O co-doping creates additional active sites and enhances the adsorption energy for electrolyte ions like K⁺, as confirmed by density functional theory (DFT) calculations [18]. This modification not only improves pseudocapacitive contributions but also enhances electrode wettability, optimizing charge carrier mobility across the graphitic lattice [18].
Pore Structure Engineering
The pore architecture of carbon materials is critical for ion transport, adsorption, and selectivity. A hierarchical structure is often ideal.
- Hierarchical Pore Design: Engineering pores across multiple scales (micro-, meso-, and macro-pores) allows each type to play a distinct role. Micropores (< 2 nm) provide numerous ion adsorption sites, directly contributing to charge storage [9] [71]. Mesopores (2-50 nm) act as ion-buffering reservoirs and low-resistance pathways, facilitating rapid ion transport to the micropores [9]. Macropores (> 50 nm) function as main channels for ion delivery from the bulk electrolyte [9]. In water-in-salt electrolytes, ions may undergo desolvation to enter sub-nanometer micropores, which can contribute more to the specific capacity than larger pores [71].
- Control of Pore Size Distribution: Studies comparing commercial activated carbons (YP50F and YP80F) with similar surface chemistry but different pore textures have shown that wider micropores facilitate faster ion dynamics [72]. The enhanced compatibility between pore size and ion steric dimensions leads to improved rate capability and frequency response in electrochemical devices [72].
Creation of Defects and Dense Structures
Beyond porosity, the density and defect concentration of the carbon matrix significantly influence volumetric performance.
- Defect Engineering: Creating defects in the carbon framework, such as through heteroatom doping, can optimize the electronic, chemical, and physical properties of the material. These defects often serve as active sites for ion adsorption and can enhance the overall electrochemical functionality [9].
- Densification for Volumetric Performance: Achieving high volumetric energy density is crucial for portable devices. A strategy involving the construction of a carbon dot-embedded amorphous carbon (CDEA) structure via electrostatic assembly and mechanical compaction has been reported [18]. This process creates a dense, three-dimensional conductive network (compaction density of 1.19 g cm⁻³) that reduces electrode impedance and provides a high density of active sites, leading to a remarkable volumetric capacitance of 373.6 F cm⁻³ [18].
Table 1: Summary of Key Functionalization Strategies and Their Impacts
| Strategy | Key Materials/Methods | Primary Effect | Performance Outcome |
|---|---|---|---|
| Surface Functionalization | Nafion-PSS composite [70] | Enhanced hydrophilicity & cation selectivity | Low detection limits for Pb²⁺ (6.478 ppb) and Cd²⁺ (5.277 ppb) |
| Heteroatom Doping | N/O-co-doping [18] | Increased active sites & improved wettability | Enhanced K⁺ adsorption energy; high gravimetric/volumetric capacitance |
| Pore Structure Engineering | Hierarchical porous carbons (HPCs) [9] | Efficient ion transport & high adsorption area | High desalination capacity & fast ion diffusion |
| Defect & Density Control | Carbon dot-embedded amorphous structure [18] | Continuous conductive pathways & high mass per volume | High volumetric capacitance (373.6 F cm⁻³) and low impedance |
Experimental Protocols and Methodologies
To validate the efficacy of the aforementioned strategies, rigorous experimental protocols for material synthesis, modification, and characterization are essential.
Synthesis of Modified Electrodes
Protocol 1: Fabrication of a Nafion-PSS Modified Screen-Printed Electrode (Nafion-PSS/SPE) [70]
- Electrode Fabrication: Manually fabricate a screen-printed electrode (SPE) using a polyester mesh screen attached to a flexible PVC sheet. Apply carbon conductive ink and use a squeegee to transfer the pattern, curing the ink at 60°C for 2 hours.
- Composite Preparation: Prepare a composite solution by mixing Nafion and poly(sodium 4-styrenesulfonate) (PSS) in a defined mass ratio in a solvent.
- Drop-Casting Modification: Deposit a precise volume of the Nafion-PSS composite solution onto the surface of the SPE working electrode via a drop-casting method.
- Drying: Allow the modified electrode to dry at room temperature, forming a stable composite film on the SPE surface.
Protocol 2: Synthesis of Nitrogen/Oxygen-Codoped Dense Porous Carbons (NDPCs) [18]
- Precursor Dispersion: Disperse coal liquefaction residue-derived carbon dots (CDs) and melamine (as a nitrogen source) in ethanol via ultrasonic treatment for 15 minutes to form a uniform solution.
- Electrostatic Assembly: Mix the CDs/melamine solution with a KOH/ethanol solution (activator) and stir magnetically for 12 hours to allow electrostatic co-assembly.
- Drying and Compaction: Dry the resulting mixture and subject it to mechanical compaction to form a dense pre-form.
- Carbonization and Activation: Carbonize the compacted pre-form at 800°C for 2 hours under a nitrogen atmosphere in a tube furnace. This one-step process simultaneously carbonizes the precursors and activates the material, yielding the final NDPC.
Characterization Techniques
A multi-technique approach is required to fully understand the structure-property relationships of modified carbon electrodes.
- Surface Wettability: Water Contact Angle (WCA) analysis is used to quantitatively evaluate hydrophilicity. A lower contact angle indicates better wettability, which is crucial for electrolyte penetration [70].
- Surface Chemistry and Morphology:
- X-ray Photoelectron Spectroscopy (XPS) determines the elemental composition and identifies the types of nitrogen and oxygen functional groups on the carbon surface [30] [18].
- Scanning Electron Microscopy (SEM) and High-Resolution Transmission Electron Microscopy (HRTEM) reveal the surface morphology, pore structure, and dense architecture of the materials [18].
- Pore Structure Analysis:
- N₂ Adsorption-Desorption Isotherms at 77 K are used to calculate the specific surface area (SBET) using the Brunauer-Emmett-Teller (BET) method and pore size distribution using Non-Local Density Functional Theory (NLDFT) [30] [18]. Parameters such as micropore volume and total pore volume are derived from this analysis.
- Electrochemical Characterization:
- Cyclic Voltammetry (CV) is employed to study the electrochemical behavior, determine the electrochemically active surface area (ECSA), and identify Faradaic processes. The ECSA for cations and anions can be separately evaluated using redox probes like [Fe(CN)₆]³⁻/⁴⁻ and [Ru(NH₃)₆]³⁺ [70].
- Electrochemical Impedance Spectroscopy (EIS) measures the charge transfer resistance and ion diffusion kinetics within the electrode structure.
Table 2: Key Performance Metrics from Cited Experimental Studies
| Material/System | Key Metric | Value | Test Conditions / Method |
|---|---|---|---|
| Nafion-PSS/SPE [70] | Detection Limit for Cd²⁺ | 5.277 ppb | Square Wave Anodic Stripping Voltammetry (SWASV) |
| Detection Limit for Pb²⁺ | 6.478 ppb | Square Wave Anodic Stripping Voltammetry (SWASV) | |
| Electrode Hydrophilicity | Reduced Water Contact Angle | Contact Angle Analysis | |
| NDPC Supercapacitor [18] | Volumetric Capacitance | 373.6 F cm⁻³ | 1 A g⁻¹ in three-electrode setup |
| Gravimetric Capacitance | 314 F g⁻¹ | 1 A g⁻¹ in three-electrode setup | |
| Compaction Density | 1.19 g cm⁻³ | - | |
| CDI for Uranium Removal [73] | U(VI) Adsorption Capacity | 680.89 mg/g | pH 4.5, N-doped GO aerogel electrode |
Visualization of Relationships and Workflows
The following diagrams illustrate the core concepts, material design workflows, and ion adsorption mechanisms discussed in this guide.
Core Challenge-Solution Framework
This diagram outlines the three primary challenges and the corresponding material-level solutions for enhancing porous carbon electrodes.
Core Challenge-Solution Framework
Electrode Fabrication and Testing Workflow
This flowchart details the experimental protocol for creating and evaluating a polymer-modified electrode for heavy metal sensing.
Polymer-Modified Electrode Workflow
Ion Adsorption and Selectivity Mechanism
This diagram illustrates the mechanisms of ion adsorption and selectivity in a hierarchically porous, functionalized carbon electrode.
Ion Adsorption and Selectivity Mechanism
The Scientist's Toolkit: Essential Research Reagents and Materials
The following table catalogues key materials and reagents critical for research in modifying porous carbon electrodes, based on the cited literature.
Table 3: Key Research Reagent Solutions for Electrode Enhancement
| Reagent/Material | Function in Research | Application Example |
|---|---|---|
| Nafion | Cation-exchange polymer; confers selectivity by allowing cation permeation while excluding anions [70]. | Heavy metal sensing (Pb²⁺, Cd²⁺); selective electrosorption. |
| Poly(sodium 4-styrenesulfonate) (PSS) | Hydrophilic polymer; enhances surface wettability and provides additional sulfonate ligands for cation binding [70]. | Improving hydrophilicity in Nafion-PSS composite films. |
| Melamine | Source of nitrogen for heteroatom doping; enhances surface polarity and creates active sites for ion adsorption [18]. | Synthesis of N-doped porous carbons for supercapacitors. |
| KOH (Potassium Hydroxide) | Chemical activator; creates micropores and increases specific surface area during carbon synthesis [74]. | Activation of sustainable porous carbons for CDI. |
| ZnCl₂ (Zinc Chloride) | Chemical activator; regulates pore structure, producing materials with a high proportion of micropores [71]. | Controlling pore size distribution in supercapacitor electrodes. |
| Carbon Dots (from Coal Liquefaction Residue) | Sustainable carbon precursor; provides structural units for creating dense, conductive 3D networks and oxygen-containing functional groups [18]. | Fabrication of high-volumetric-performance dense porous carbons. |
The challenges of surface activity, hydrophilicity, and selectivity in porous carbon electrodes are interconnected but addressable through deliberate material design. The integration of polymer composites like Nafion-PSS, strategic heteroatom doping, and the engineering of hierarchical pore structures represent proven pathways to significant performance enhancement. The experimental protocols and characterization techniques outlined provide a roadmap for researchers to validate new material designs. As the field progresses, the fusion of these strategies—such as creating dense, defect-rich, N/O-codoped carbons with tailored surface chemistry—will be pivotal in advancing the capabilities of electrochemical systems for energy storage and selective ion separation. The continued refinement of these approaches, guided by robust theoretical and experimental insights, will unlock new possibilities in environmental remediation and resource recovery.
Validating Performance Through Comparative Analysis and Advanced Characterization
The escalating challenges of environmental pollution and the urgent need for sustainable water and air purification technologies have propelled research into advanced adsorbent materials. Within this context, porous carbon electrodes and their adsorption capabilities have become a focal point of scientific inquiry. This whitepaper examines the comparative adsorption performance of biomass-derived porous carbons against traditional adsorbent materials, framing the analysis within the broader research on ion adsorption in porous carbon electrodes. The drive towards sustainable material sourcing, coupled with the demand for high-efficiency adsorption, positions biomass carbons as a compelling subject for investigation by researchers and scientists engaged in environmental technology and material development.
Theoretical Framework: Adsorption in Porous Carbon Materials
The adsorption capacity of porous carbon materials is governed by a complex interplay of physical structure and surface chemistry. Biomass-derived activated carbons function through a multifaceted adsorption mechanism that includes physical adsorption via van der Waals forces in their porous networks, electrostatic interactions for charged species, and chemical adsorption through surface functional groups [75] [76].
Recent theoretical advances propose an entropy-driven framework for designing high-performance carbon materials. The concept of "high-entropy carbon" introduces three distinct pathways for enhancing system disorder: unit entropy (decreasing graphene domain size to increase basic units), ring entropy (distorting ideal six-membered carbon rings to create topological defects), and element entropy (doping multiple heteroatoms into the graphene lattice) [3]. This entropy-driven approach increases the diversity of adsorption sites, potentially leading to novel capacitance storage mechanisms and enhanced ion adsorption capabilities.
The efficacy of these materials is quantified through their specific surface area (SSA), pore size distribution, and surface functional groups, which collectively determine their accessibility and affinity for target adsorbates [9] [75]. The presence of heteroatoms such as nitrogen and oxygen creates electron-donor/acceptor sites that significantly enhance adsorption of various contaminants through improved charge distribution [18].
Biomass-Derived Porous Carbons: Synthesis and Characteristics
Precursor Materials and Sustainable Sourcing
Biomass-derived activated carbons are synthesized from abundant and renewable agricultural wastes, representing a sustainable approach to material fabrication. Common precursors include:
- Coconut shells: Produce activated carbon with specific surface area exceeding 516 m²/g when activated with NaOH [77]
- Bamboo: Characterized by inherent high potassium content and narrow fibrous channels that enhance selective CO₂ adsorption [78]
- Lignin: Yields porous carbon with layered graphene-like structure ideal for heavy metal immobilization [79]
- Agricultural wastes: Including rice husks, coconut shells, and other byproducts that contribute to waste reduction while creating valuable adsorbents [75] [76]
The utilization of these materials aligns with circular economy principles, transforming waste products into functional materials with significant environmental benefits [9] [76].
Synthesis and Activation Methodologies
The fabrication of biomass-derived activated carbons follows a systematic protocol to develop optimal porosity and surface functionality:
Figure 1: Synthesis workflow for biomass-derived activated carbons, highlighting key stages from precursor selection to functionalized material production.
Carbonization Process: Biomass precursors undergo thermal decomposition at temperatures ranging from 600°C to 800°C under inert nitrogen atmosphere for 1-3 hours, preventing complete combustion of lignin and cellulose components [78] [80]. This process removes volatile components and creates the initial carbon framework.
Activation Methods:
- Chemical Activation: Treatment with activating agents including KOH, NaOH, H₃PO₄, or ZnCl₂ at specific mass ratios (typically 1:1 to 1:5.5 char:activator) followed by pyrolysis at 700-800°C [77] [80]. NaOH activation has been shown to achieve surface areas of 516 m²/g for coconut shell-based carbons [77].
- Physical Activation: Exposure to steam or CO₂ at elevated temperatures (800°C for 2 hours) to develop porosity through gasification reactions [78].
Post-Synthesis Modifications:
- Amination: Treatment with ammonia gas at 800°C to introduce nitrogen-containing functional groups (pyridine, pyrrole, quaternary N) that enhance CO₂ selectivity [78].
- Magnetic Modification: Impregnation with FeCl₃ solutions (25-100 mmol/L) followed by pyrolysis to create easily separable adsorbents [80].
- Heteroatom Doping: Introduction of nitrogen and oxygen functional groups to enhance surface polarity and electron density [18].
Performance Comparison: Quantitative Analysis
Adsorption Capacity for Heavy Metals
Biomass-derived activated carbons demonstrate exceptional performance in heavy metal removal from aqueous solutions, outperforming many conventional adsorbents.
Table 1: Heavy Metal Adsorption Capacity of Biomass-Derived Activated Carbons
| Adsorbent Material | Precursor | Target Contaminant | Adsorption Capacity (mg/g) | Reference |
|---|---|---|---|---|
| Lignin-based Porous Carbon | Lignin | Pb(II) | 250.5 | [79] |
| Lignin-based Porous Carbon | Lignin | Cd(II) | 126.4 | [79] |
| Magnetic Biochar | Coconut Shell | Sulfadiazine | 294.12 | [80] |
| Magnetic Biochar | Coconut Shell | Sulfamethazine | 400.00 | [80] |
| Magnetic Biochar | Coconut Shell | Sulfamethoxazole | 454.55 | [80] |
| KOH-Activated Carbon | Coconut Shell | Various wastewater contaminants | High surface area: 516 m²/g | [77] |
The superior adsorption capacities observed in biomass-derived carbons are attributed to their tailored surface chemistry and hierarchical pore structures that facilitate both diffusion and immobilization of heavy metal ions [79] [75]. The presence of oxygen-rich functional groups (-OH, -COOH) enables complexation with metal ions, while the porous structure provides numerous adsorption sites.
Gaseous Contaminant Adsorption
The performance of biomass carbons in gaseous contaminant removal demonstrates their versatility across different environmental applications.
Table 2: Gaseous Contaminant Adsorption Performance
| Adsorbent Material | Precursor | Target Contaminant | Adsorption Capacity | Conditions | Reference |
|---|---|---|---|---|---|
| Aminated Bamboo AC | Bamboo | CO₂ (0.3%) | Selectivity (αs,g): 13.4 | Ambient | [78] |
| Bamboo AC | Bamboo | CO₂ (pure) | 1.80 mmol/g | Ambient | [78] |
| Bamboo AC | Bamboo | CO₂ (3000 ppm) | 0.98 mmol/g | Ambient | [78] |
| Commercial Coconut AC | Coconut Shell | CO₂ (0.3%) | Selectivity (αs,g): 1.16-1.38 | Ambient | [78] |
| N/O-doped Dense Carbon | Coal residue | K⁺ ions | Enhanced adsorption energy | Electrochemical system | [18] |
Bamboo-derived activated carbon exhibits particular promise for CO₂ capture, achieving significantly higher selectivity compared to commercial coconut shell-based alternatives [78]. This enhanced performance is attributed to bamboo's inherent high potassium content and the development of predominantly microporous structures with an average pore size of 0.17 nm, which is ideal for capturing CO₂ molecules [78].
Comparison with Traditional Adsorbents
When evaluated against conventional adsorption materials, biomass-derived carbons demonstrate competitive or superior performance in multiple applications:
Compared to Metal-Organic Frameworks (MOFs):
- Biomass carbons exhibit superior moisture resistance and greater structural stability under varying environmental conditions [81]
- While some MOFs achieve higher surface areas (up to 15,000 m²/g), their selective adsorption capacity for gaseous separation is often lower than functionalized biomass carbons [78]
- Biomass carbons offer significant economic advantages, with production costs substantially lower than synthesized MOFs [9]
Compared to Zeolites:
- Biomass carbons maintain adsorption efficiency in humid conditions, whereas zeolites exhibit sensitivity to moisture [78]
- Aminated biomass carbons demonstrate CO₂ selectivity factors (13.4) that surpass many zeolitic materials [78]
Compared to Traditional Activated Carbons:
- Biomass-derived variants offer sustainable sourcing advantages over coal-based activated carbons [9]
- Tailored surface functionality through heteroatom doping provides enhanced selectivity for specific contaminants [18]
- Agricultural waste precursors reduce material costs while maintaining competitive performance metrics [75]
Advanced Material Engineering Strategies
Defect and Structure Engineering
Contemporary research focuses on deliberate engineering of carbon structures at multiple scales to enhance adsorption performance:
Defect Engineering: Introduction of topological defects through entropy-driven design principles, including five- and seven-membered carbon rings that create charge localization sites for enhanced ion adsorption [3].
Heteroatom Doping: Incorporation of nitrogen and oxygen functional groups significantly enhances pseudocapacitive behavior and ion adsorption capacity. Experimental results demonstrate that N/O co-doping increases K⁺ adsorption energy, with optimized materials achieving volumetric capacitances of 373.6 F cm⁻³ [18].
Dense Porous Architecture: Development of compact carbon structures with high packing density (1.19 g cm⁻³) that provide continuous conductive pathways while maintaining accessible porosity. This approach addresses the volumetric capacity limitations of conventional high-surface-area carbons [18].
Sustainable Activation Approaches
Recent advances focus on addressing environmental concerns associated with traditional activation methods:
Green Activators: Development of inorganic salt and organic salt activators as alternatives to corrosive chemicals like KOH and HF, reducing environmental impact while maintaining performance [9].
Sustainable Manufacturing: Implementation of closed-loop systems that minimize waste generation and energy consumption during carbon production [9].
Magnetic Modification: Facilitation of adsorbent recovery and reuse through iron oxide incorporation, enabling multiple adsorption-desorption cycles with minimal capacity loss [80].
Experimental Protocols and Methodologies
Standardized Testing Procedures
To ensure reproducible evaluation of adsorption performance, researchers employ standardized experimental protocols:
Aqueous Phase Adsorption Testing:
- Prepare contaminant solutions at varying concentrations (e.g., 10-500 mg/L for heavy metals)
- Adjust solution pH using HNO₃ or NaOH to examine pH-dependent adsorption behavior
- Add fixed mass of adsorbent (e.g., 0.1 g) to solution volume (e.g., 100 mL)
- Agitate mixture at constant temperature (e.g., 25°C) for specified duration (typically 24 hours for equilibrium)
- Filter and analyze supernatant using appropriate analytical techniques (AAS, ICP-MS, UPLC)
- Calculate adsorption capacity using: qₑ = (C₀ - Cₑ) × V / m, where qₑ is equilibrium adsorption capacity, C₀ and Cₑ are initial and equilibrium concentrations, V is solution volume, and m is adsorbent mass [79] [75] [80]
Gaseous Phase Adsorption Testing:
- Utilize fixed-bed adsorption columns with precise temperature control
- Introduce gas mixtures at controlled flow rates and concentrations (e.g., 0.3% CO₂ in N₂ for indoor air simulations)
- Monitor breakthrough curves using gas analyzers or GC systems
- Calculate adsorption capacity from integrated breakthrough curves [78] [76]
Regeneration and Reusability Assessment:
- Perform desorption using appropriate eluents (acids for metals, temperature swing for gases)
- Conduct multiple adsorption-desorption cycles (typically 3-5 cycles)
- Measure capacity retention after each cycle to assess long-term viability [79] [80]
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Materials and Reagents for Biomass Carbon Research
| Reagent/Material | Function | Application Example | Reference |
|---|---|---|---|
| KOH / NaOH | Chemical activation | Porosity development in coconut shell and bamboo carbons | [77] [80] |
| ZnCl₂ / H₃PO₄ | Chemical activation | Alternative activation agents for specific pore structures | [77] [81] |
| NH₃ gas | Surface amination | Introduction of nitrogen functionalities for enhanced CO₂ capture | [78] |
| FeCl₃·6H₂O | Magnetic modification | Impregnation for magnetic separation and recovery | [80] |
| HNO₃ | Surface oxidation | Introduction of oxygen-containing functional groups | [80] |
| Melamine | Nitrogen doping | Source of nitrogen for heteroatom doping | [18] |
Biomass-derived activated carbons demonstrate competitive, and in many cases superior, adsorption capacity compared to traditional adsorbent materials across diverse applications including heavy metal removal, CO₂ capture, and organic contaminant adsorption. Their sustainable sourcing, tunable surface chemistry, and hierarchical porous structures position them as compelling alternatives to conventional materials.
The integration of entropy-driven design principles [3] with advanced functionalization techniques [78] [18] has enabled precise engineering of biomass carbons with enhanced ion adsorption capabilities. The exceptional performance of these materials in both aqueous and gaseous phase applications underscores their potential to address pressing environmental challenges.
Future research should focus on scaling sustainable fabrication methods [9], advancing our understanding of molecular-level adsorption mechanisms [3] [18], and developing standardized testing protocols for reliable performance comparison. As the field progresses, biomass-derived porous carbons are poised to play an increasingly significant role in environmental remediation, energy storage, and sustainable technological development.
In Situ NMR and Spectroscopic Validation of Adsorption Mechanisms
Understanding ion adsorption mechanisms in porous carbon electrodes is a fundamental challenge in advancing energy storage technologies such as supercapacitors and batteries. The complex pore architectures, diverse ion desolvation states, and dynamic interfacial phenomena occurring during electrochemical operation necessitate advanced characterization techniques that can provide molecular-level insights under working conditions. Among these techniques, in situ Nuclear Magnetic Resonance (NMR) spectroscopy has emerged as a powerful tool for probing local environments, ion dynamics, and charge storage mechanisms with exceptional specificity. This technical guide explores how in situ NMR and complementary spectroscopic methods provide direct experimental validation of adsorption mechanisms, framed within the broader context of ion adsorption research in porous carbon electrodes.
The significance of this approach lies in its ability to resolve long-standing debates in the field, including the relative contributions of ion reorganization versus electronic effects, the nature of ion desolvation in confined pores, and the molecular origins of anomalous phenomena such as oversolubility. By integrating in situ NMR observations with computational modeling and electrochemical measurements, researchers can establish definitive structure-property relationships that guide the rational design of next-generation energy storage materials.
Fundamental Principles of In Situ NMR for Adsorption Studies
Ring Current Effects and Chemical Shift
The ring current effect represents a fundamental principle underpinning NMR studies of carbon-based electrodes. When aromatic carbon structures (such as graphene domains) experience an external magnetic field, their delocalized π-electrons generate circulating ring currents. These currents, in turn, produce secondary magnetic fields that significantly influence the resonant frequency (chemical shift) of nuclei in proximity. During electrochemical polarization, changes in electronic density within the carbon electrode alter these ring currents, leading to measurable chemical shift variations that provide insights into electronic structure modifications during charging and discharging [82] [83].
Research has demonstrated that ring currents constitute the dominant contribution to observed chemical shift variations in supercapacitor electrodes, with ion reorganization playing a secondary role. This understanding resolves previous ambiguities in interpreting in situ NMR spectra and establishes a foundation for correlating electronic structure with capacitive performance [82].
Probing Ion Environments and Dynamics
In situ NMR enables the discrimination between different ionic states within operating electrochemical devices. Through chemical shift analysis, researchers can distinguish between:
- Free ions in the bulk electrolyte
- Adsorbed ions within porous carbon electrodes
- Ions in different confinement environments based on pore size
This discrimination capability proves particularly valuable for studying ion desolvation states, which significantly influence charge storage mechanisms. For example, potassium ions in porous carbons can exist in five distinct desolvation states, denoted as [K(H₂O)₀₋₄]⁺, each with characteristic energetics and dynamics [15]. Similar principles apply to organic electrolytes and ionic liquids, where ion pairing and coordination states affect capacitive behavior.
Table 1: Key NMR-Active Nuclei for Studying Adsorption Mechanisms in Energy Storage Materials
| Nucleus | Isotopic Abundance | Relevant Applications | Information Obtained |
|---|---|---|---|
| ¹³C | 1.1% | CO₂ adsorption/oversolubility studies, carbon structure analysis | Chemical identity, adsorption configuration, pore confinement effects [84] [85] |
| ¹H | 99.9% | Solvent organization, ion solvation/desolvation, water dynamics | Molecular mobility, chemical environment, intermolecular interactions [15] [83] |
| ¹⁹F | 100% | PF₆⁻ anion tracking, ionic liquid studies | Ion adsorption, coordination changes, mobility in confinement [83] |
| ¹¹B | 80.4% | BF₄⁻ anion behavior in ionic liquids | Ion partitioning, adsorption preferences, dynamics [83] |
| ⁷Li | 92.4% | Lithium-ion battery systems | Ion intercalation, solvation structure, diffusion mechanisms [83] |
Experimental Methodologies and Protocols
In Situ NMR Cell Design
Implementing in situ NMR requires specialized electrochemical cells compatible with NMR instrumentation while maintaining controlled electrochemical conditions:
- Capillary-based cells utilizing glass or polyether ether ketone (PEEK) materials that minimize magnetic susceptibility distortions
- Three-electrode configuration with working electrode (porous carbon material), reference electrode, and counter electrode
- Metallic current collectors optimized to minimize NMR signal interference
- Electrolyte reservoir ensuring sufficient ion supply during operation
- Magic Angle Spinning (MAS) capabilities for high-resolution studies, though static measurements are more common for in situ experiments
Core NMR Techniques for Adsorption Studies
Chemical Shift Mapping
Protocol:
- Acquire NMR spectra at progressively increasing electrode potentials
- Reference chemical shifts to a standard compound
- Correlate shift changes with applied potential
- Deconvolute contributions from ring currents versus ion reorganization
Application: This approach revealed that ring current contributions dominate chemical shift variations in porous carbon supercapacitors, accounting for approximately 70-80% of the observed shift, while ion reorganization contributes the remainder [82].
Pulsed Field Gradient (PFG) NMR
Protocol:
- Apply matched magnetic field gradients to encode spatial position
- Measure signal attenuation as a function of gradient strength
- Calculate diffusion coefficients using the Stejskal-Tanner equation
- Compare diffusion rates in bulk versus confined environments
Application: PFG-NMR quantifies ion mobility within porous carbon electrodes, revealing restricted diffusion in micropores (<2 nm) versus nearly bulk-like mobility in macropores (>50 nm) [83].
Magic Angle Spinning (MAS) with Cross Polarization (CP)
Protocol:
- Spin sample at the magic angle (54.74°) to average anisotropic interactions
- Transfer polarization from abundant nuclei (¹H) to rare nuclei (¹³C)
- Enhance sensitivity for low-γ nuclei
- Resolve distinct chemical environments
Application: ¹³C CP-MAS NMR identified six distinct adsorbed CO₂ species (three chemisorbed, three physisorbed) in amine-modified porous silica, enabling individual adsorption isotherm construction for each species [85].
Remote Detection NMR
Protocol:
- Encode spatial/spectral information within the porous material using a large detection coil
- Transfer the encoded spin ensemble to a detection region via fluid flow
- Detect signal with a small, optimized coil outside the porous material
- Reconstruct information using time-of-flight correlation
Application: This method provided 300-700-fold sensitivity enhancement for quantifying propane/propene adsorption in mesoporous materials under continuous flow conditions [86].
The following diagram illustrates the typical workflow for an in situ NMR study of adsorption mechanisms:
Diagram 1: Experimental workflow for in situ NMR adsorption studies
Key Insights into Adsorption Mechanisms
Ion Desolvation States in Aqueous Systems
In situ NMR studies have revealed the complex desolvation behavior of ions entering subnanometer pores in carbon electrodes. For potassium ions in aqueous electrolytes, five distinct desolvation states ([K(H₂O)₀₋₄]⁺) with different adsorption energies and diffusion barriers have been identified [15]. The distribution among these states depends critically on pore size and surface chemistry, with oxygen functional groups significantly influencing the preferred desolvation pathway.
The quantitative understanding of desolvation thermodynamics and kinetics enables dual optimization strategies targeting both thermodynamic stabilization of partial desolvation states and kinetic enhancement of ion transport. This approach has led to specific capacitances as high as 273 F g⁻¹ in coal-derived porous carbon electrodes [15].
Oversolubility Phenomena in Microporous Carbons
A remarkable phenomenon uncovered through in situ NMR is the oversolubility of gases in solvent-saturated microporous carbons. Using ¹³C NMR spectroscopy, researchers observed up to a 30-fold enhancement of CO₂ solubility in microporous activated carbons completely saturated with aqueous electrolyte [84].
This oversolubility effect occurs through an adsorption-like mechanism driven by favorable CO₂-pore wall interactions that outcompete solvent-pore wall interactions. The effect is enhanced in smaller pores and is largely independent of the carbon's functional groups or degree of structural disorder [84]. This phenomenon has significant implications for electrochemical CO₂ capture and reduction systems, where local CO₂ concentration at the electrode-electrolyte interface critically determines performance.
Ion Storage Mechanisms in Ionic Liquids
For ionic liquid electrolytes such as [EMIM][BF₄], in situ NMR has helped elucidate the pore-size-dependent ion adsorption behavior. In pores larger than 1.3 nm, anions and cations adsorb alternately on carbon walls due to Coulombic ordering. As pore size decreases to 0.9-1.3 nm, this ordering breaks down, giving way to a "double-layer ionic monotonic adsorption" mechanism where cations ([EMIM]⁺) adsorb in a parallel arrangement on both pore walls [87].
This mechanism maximizes charge storage density while maintaining rapid ion transport, enabling supercapacitors with exceptional energy density (109 Wh kg⁻¹) and power density (71 kW kg⁻¹) [87].
Table 2: Quantitative Insights Gained from In Situ NMR Studies of Porous Carbon Electrodes
| Phenomenon | System | NMR Technique | Key Quantitative Finding | Reference |
|---|---|---|---|---|
| Ion desolvation | K⁺ in porous carbon | ¹H/³⁹K NMR | Five distinct desolvation states [K(H₂O)₀₋₄]⁺ with desolvation energies from 0.5-1.8 eV | [15] |
| CO₂ oversolubility | Activated carbon/Na₂SO₄(aq) | ¹³C MAS NMR | 30-fold solubility enhancement in micropores; 5-20× enhancement from dissolved CO₂ alone | [84] |
| Ring current effects | Porous carbon/organic electrolyte | In situ ¹⁹F/¹¹B NMR | Ring currents account for ~80% of chemical shift variation during polarization | [82] |
| Multi-component adsorption | Controlled pore glass/C₃H₆-C₃H₈ | Remote detection NMR | 300-700× sensitivity enhancement enabling flow adsorption quantification | [86] |
| Ionic liquid adsorption | 3DPFC/[EMIM][BF₄] | In situ NMR | "Double-layer ionic monotonic adsorption" in 0.9-1.3 nm pores enables 109 Wh kg⁻¹ | [87] |
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Research Reagent Solutions for In Situ NMR Studies of Adsorption Mechanisms
| Reagent/Material | Function in Research | Specific Application Example |
|---|---|---|
| ¹³C-enriched CO₂ | Tracer for adsorption pathway studies | Quantifying oversolubility mechanisms in microporous carbons [84] [85] |
| Ionic liquids (e.g., [EMIM][BF₄]) | High-voltage electrolyte for supercapacitors | Studying ion adsorption mechanisms in confined pores [87] |
| Paramagnetic relaxation agents | Differentiating adsorbed vs. bulk species | Probing ion partitioning in porous carbon electrodes [83] |
| Controlled pore glass materials | Model porous systems with well-defined porosity | Validating NMR methods for adsorption quantification [86] |
| Isotopically labeled electrolytes | Enhancing NMR sensitivity for specific ions | Tracking ion desolvation states in aqueous systems [15] |
| Amine-modified mesoporous silica | Reference sorbent for CO₂ capture studies | Distinguishing six individual CO₂ adsorption species [85] |
| Magic Angle Spinning (MAS) rotors | Enabling high-resolution NMR of heterogeneous samples | Resolving distinct chemical environments in porous materials [85] |
Integration with Computational Methods
The interpretation of in situ NMR data is greatly enhanced through integration with computational approaches. Mesoscopic simulations have been particularly valuable for decoupling the relative contributions of ring current effects versus ion reorganization to observed chemical shifts [82]. These simulations achieve quantitative agreement with experimental NMR spectra, confirming that ring current effects dominate the potential-dependent chemical shift variations.
Atomistic modeling based on machine-learning potentials has provided molecular-level insights into oversolubility phenomena, revealing that favorable CO₂-pore wall interactions drive the observed solubility enhancements in microporous carbons [84]. Similarly, Density Functional Theory (DFT) and Molecular Dynamics (MD) simulations have elucidated the relationship between pore dimensionality and ion storage kinetics, demonstrating enhanced ion adsorption energies and reduced diffusion barriers in 2D porous carbons compared to 3D architectures [88].
The following diagram illustrates the complementary relationship between experimental NMR observations and computational modeling in validating adsorption mechanisms:
Diagram 2: Integration of NMR and computational methods
In situ NMR spectroscopy has transformed our understanding of adsorption mechanisms in porous carbon electrodes, moving beyond macroscopic descriptions to molecular-level mechanistic insights. The technique has successfully resolved long-standing questions regarding the relative importance of electronic versus ionic contributions to capacitive behavior, revealed the complexity of ion desolvation states in nanoconfinement, and uncovered unexpected phenomena such as gas oversolubility in solvent-saturated micropores.
Future developments in this field will likely focus on enhancing temporal resolution to capture dynamic processes at relevant timescales, improving spatial resolution through advanced imaging techniques, and expanding the range of nuclei and conditions accessible to in situ investigation. Combining NMR with complementary techniques such as X-ray scattering, vibrational spectroscopy, and electrochemical quartz crystal microbalance measurements will provide multidimensional insights into adsorption processes.
As porous carbon materials continue to evolve in complexity—incorporating heteroatom doping, multimodal porosity, and tailored surface chemistry—in situ NMR will remain an indispensable tool for validating adsorption mechanisms and guiding the rational design of next-generation energy storage systems. The methodology provides an essential bridge between theoretical predictions and experimental validation, ensuring that fundamental insights translate into practical material advances.
The performance of porous carbon electrodes in ion adsorption is critically defined by their maximum adsorption capacity, a key benchmark for evaluating and comparing material efficacy in both research and industrial applications. Within the broader context of ion adsorption research, achieving high capacity is paramount for developing efficient environmental remediation and resource recovery technologies. This guide provides a structured approach to analyzing these performance benchmarks, consolidating current data and methodologies to establish a clear framework for assessing carbon-based electrodes. The focus is on creating a standardized reference that enables direct comparison across different material modifications and target ions, supported by quantitative data and detailed experimental protocols.
Quantitative Benchmarks for Ion Adsorption Capacities
Table 1: Maximum Adsorption Capacities of Carbon-Based Electrodes for Target Ions
| Target Ion | Electrode Material | Experimental Conditions | Maximum Adsorption Capacity | Reference |
|---|---|---|---|---|
| U(VI) | Nitrogen-doped GO Aerogel | Not Specified | 680.89 mg/g | [73] |
| U(VI) | Malonamide–Amidoxime-functionalized GO | pH 4.5 | 479.4 mg/g | [73] |
| U(VI) | Graphene Oxide/Polypyrrole Hybrid Film | Applied voltage: 1.2 V | 301.0 mg/g | [73] |
| Maleic Acid | Activated Carbon Cloth (ACC) | Applied potential: +1.00 V vs. Ag/AgCl | 50.40 mg/g | [89] |
The data demonstrates that graphene oxide (GO)-based materials, particularly through advanced functionalization and doping, achieve the highest recorded capacities for uranium removal. The progression from functionalized GO (479.4 mg/g) to nitrogen-doped GO aerogels (680.89 mg/g) highlights the significant performance gains possible through strategic material design [73]. Furthermore, the application of an electrical potential is a critical factor, as evidenced by the more than five-fold improvement in maleic acid uptake on Activated Carbon Cloth (ACC) at +1.00 V compared to open-circuit conditions [89].
Detailed Experimental Protocols for Benchmarking
To ensure the reproducibility and reliability of adsorption capacity data, adherence to detailed experimental protocols is essential. The following methodologies are derived from cited research.
Electrosorption Experiment Protocol for Uranium
This protocol outlines the general procedure for evaluating the electrosorption capacity of carbon-based electrodes for uranium, as described in studies on graphene oxide and functionalized materials [73].
- Electrode Preparation: Fabricate the carbon-based working electrode (e.g., through electrodeposition of graphene oxide on carbon felt or preparing a free-standing ACC). Use an Ag/AgCl reference electrode and a platinum counter electrode to complete the circuit [73].
- Solution Preparation: Prepare a solution containing the target ion (U(VI)) at a specific initial concentration in a controlled background electrolyte. The pH should be adjusted to a predetermined value using HCl or NaOH [73] [89].
- Applied Potential: Introduce the electrodes into the solution and apply a constant voltage (e.g., 1.2 V) across the electrode pair using a potentiostat [73].
- Kinetic Sampling: Operate the system for a set duration, collecting solution samples at regular intervals to monitor the change in ion concentration over time.
- Analysis and Calculation: Measure the uranium concentration in the samples using inductively coupled plasma mass spectrometry (ICP-MS) or another suitable analytical technique. The adsorption capacity at time t, q_t (mg/g), is calculated as: q_t = (C_0 - C_t) * V / m where C_0 and C_t are the initial and at-time-t concentrations (mg/L), V is the solution volume (L), and m is the mass of the adsorbent (g) [73].
Protocol for Adsorption-Desorption Cycling
Assessing the regenerability and stability of electrodes is crucial for practical applications. This protocol is adapted from work on Activated Carbon Cloth [89].
- Adsorption Step: Follow the electrosorption protocol (Steps 1-4) at a positive potential (e.g., +1.00 V vs. Ag/AgCl) to load the target ion onto the electrode.
- Desorption Step: After reaching adsorption equilibrium, switch the applied potential to a negative value (e.g., -1.00 V vs. Ag/AgCl) or switch to open-circuit potential in a clean solution without the target ion.
- Regeneration Measurement: Monitor the concentration of the target ion released into the solution during desorption to calculate the recovery efficiency.
- Cycle Repetition: Repeat the adsorption-desorption steps multiple times to evaluate the electrode's capacity retention and long-term stability [89].
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Materials and Reagents for Ion Adsorption Experiments
| Item | Function/Description | Example Use Case |
|---|---|---|
| Activated Carbon Cloth (ACC) | A free-standing, binder-free porous carbon electrode with high specific surface area, facilitating excellent ion and electron transport. | Serves as the primary electrode material for the electrosorption of organic acids like maleic acid [89]. |
| Graphene Oxide (GO) | A carbon nanomaterial with a tunable surface rich in oxygen-containing functional groups, which can be further modified to enhance selectivity and capacity. | Used as a base material for creating high-capacity electrodes for U(VI) removal; can be functionalized with malonamide-amidoxime groups [73]. |
| Ag/AgCl Reference Electrode | Provides a stable and reproducible reference potential for electrochemical measurements in a three-electrode setup. | Used to accurately control the potential applied to the working electrode during electrosorption experiments [89]. |
| Potentiostat/Galvanostat | An electronic instrument that controls the voltage (potentiostat) or current (galvanostat) between the working and counter electrodes. | Used to apply precise potentials during chronoamperometry or cyclic voltammetry experiments [89]. |
| NaCl Background Electrolyte | An inert salt used to create a controlled ionic strength environment, which influences the formation of the electrochemical double layer. | Studied at concentrations from 0-20 mM to understand its competitive effect on maleic acid uptake [89]. |
Visualization of Ion Adsorption Mechanisms
The high adsorption capacities of advanced carbon electrodes are achieved through several synergistic mechanisms, which can be engineered via material design.
The mechanisms show that performance gains are not achieved through a single strategy but through the integration of Electric Double Layer (EDL) formation, surface complexation, and pseudo-capacitive reactions [73]. This multi-mechanism approach is critical for developing next-generation electrodes that are both high-capacity and highly selective for specific target ions like U(VI).
In the field of porous carbon electrodes research, the adsorption of ions is a fundamental process with critical applications in environmental remediation, energy storage, and bioinspired iontronic devices. A comprehensive thermodynamic validation of this process is indispensable for optimizing material design and predicting system behavior. This whitepaper provides an in-depth technical guide on validating the spontaneity (through Gibbs free energy, ΔG°), enthalpy (ΔH°), and activation energy of ion adsorption onto porous carbon materials. Framed within a broader thesis on ion adsorption mechanisms, this guide consolidates experimental protocols, quantitative data, and visualization tools to support researchers and scientists in the rigorous characterization of these key thermodynamic parameters. Understanding these fundamentals is vital for advancing technologies in supercapacitor energy storage, capacitive deionization, and heavy metal removal from wastewater [10] [90] [91].
Core Thermodynamic Principles in Ion Adsorption
Ion adsorption in porous carbons is a complex process governed by the interplay of several thermodynamic factors. The Gibbs free energy change (ΔG°) indicates the spontaneity of the adsorption process; a negative value confirms a thermodynamically favorable, spontaneous reaction. The enthalpy change (ΔH°) reveals whether the process is exothermic (heat-releasing) or endothermic (heat-absorbing), providing insight into the nature of the adsorbate-adsorbent interaction. Finally, the activation energy (Ea) represents the minimum energy barrier that must be overcome for the adsorption to occur, directly influencing the kinetics and rate of the process [92] [91].
The spontaneity of adsorption is profoundly influenced by the properties of the electrolyte and the carbon material. Research has demonstrated that ion solvation energy and confinement effects within carbon micropores directly impact the spontaneous distribution of ions. Systems can be classified as ionophilic (spontaneously favoring ion adsorption) or ionophobic (spontaneously resisting ion adsorption), which in turn dictates the charge-balancing mechanism under an applied potential in supercapacitors [10]. Furthermore, the presence of ionic impurities in porous carbons can significantly alter the capacitive behavior and must be accounted for in thermodynamic analyses [93].
Quantitative Thermodynamic Data
The following tables summarize key thermodynamic parameters reported from various studies on ion and molecule adsorption using porous carbon materials.
Table 1: Thermodynamic Parameters for Organic Dye Adsorption on Activated Carbons from Green Tea Residues [92]
| Adsorbate | ΔG° (kJ/mol) | ΔH° (kJ/mol) | ΔS° (J/mol·K) | Adsorption Capacity (mg/g) |
|---|---|---|---|---|
| Methylene Blue | Not Specified | Not Specified | Not Specified | 144.93 - 250.00 |
| Methyl Red | Negative Values Reported | Not Specified | Not Specified | Not Specified |
Table 2: Thermodynamic Parameters for Heavy Metal Adsorption on Biomass-Derived Activated Carbons
| Carbon Source | Adsorbate | ΔG° (kJ/mol) | ΔH° (kJ/mol) | ΔS° (J/mol·K) | Reference |
|---|---|---|---|---|---|
| Chickpea Husks | Pb(II), Cr(VI), Cu(II) | Negative | Endothermic (Positive) | Not Specified | [91] |
| Hard Wood Composite | Pb(II) | Negative | Endothermic | Not Specified | [94] |
| Hard Wood Composite | Cd(II) | Negative | Endothermic | Not Specified | [94] |
| Winemaking Waste | Cu(II) | Not Specified | Not Specified | Not Specified | [95] |
Table 3: Activation Energy and Kinetic Data for Ion Adsorption
| System / Material | Activation Energy (Ea) | Kinetic Model | Key Finding | Reference |
|---|---|---|---|---|
| Microporous Carbon Electrodes | Determined via Multiscale Model | Pseudo-First-Order, Butler-Volmer | Ion hydration & pore size dictate Ea | [90] |
| Winemaking Waste Carbon | Physisorption (Low Ea) | Pseudo-Second-Order | Ea suggests physisorption | [95] |
| Chickpea Husk AC | Not Specified | Pseudo-Second-Order | - | [91] |
| Green Tea Residue AC | Not Specified | Pseudo-Second-Order | - | [92] |
Experimental Protocols for Thermodynamic Validation
Batch Adsorption Studies for ΔG°, ΔH°, and ΔS°
Objective: To determine the spontaneity (ΔG°), enthalpy (ΔH°), and entropy (ΔS°) changes of the adsorption process through equilibrium experiments at different temperatures [91].
Materials:
- Adsorbents: Porous carbon material (e.g., activated carbon from chickpea husks, green tea residues, or hard wood composite).
- Adsorbate Solutions: Standard solutions of the target ion (e.g., Pb(II), Cu(II), methylene blue) at known concentrations.
- Equipment: Thermostatic mechanical shaker, centrifuge, atomic absorption spectrometer (AAS) or UV-Vis spectrophotometer, pH meter.
Procedure:
- Solution Preparation: Prepare a series of adsorbate solutions with varying initial concentrations (e.g., 10-400 mg/L) using stock solutions.
- pH Adjustment: Adjust the pH of each solution to the desired value using 0.1 M NaOH or HCl.
- Equilibrium Adsorption: In sealed Erlenmeyer flasks, add a fixed mass of the porous carbon adsorbent to a fixed volume of each adsorbate solution.
- Temperature Variation: Place the flasks in a thermostatic mechanical shaker and agitate at a constant speed until equilibrium is reached. Repeat this entire process at different temperatures (e.g., 20°C, 30°C, 40°C).
- Analysis: After agitation, separate the adsorbent by filtration or centrifugation. Analyze the equilibrium concentration of the adsorbate in the supernatant using AAS (for metals) or UV-Vis spectrophotometry (for dyes).
- Data Calculation: The amount adsorbed at equilibrium, ( qe ) (mg/g), is calculated as: ( qe = \frac{(C0 - Ce)}{m} \cdot V ) where ( C0 ) and ( Ce ) are the initial and equilibrium concentrations (mg/L), ( V ) is the solution volume (L), and ( m ) is the adsorbent mass (g) [92] [91].
Thermodynamic Calculations:
- The equilibrium constant, ( K_c ), can be determined from the adsorption data at different temperatures.
- Gibbs Free Energy (ΔG°) is calculated using: ( \Delta G^\circ = -RT \ln K_c ), where ( R ) is the universal gas constant and ( T ) is the temperature in Kelvin. A negative ΔG° confirms a spontaneous process [92].
- Enthalpy (ΔH°) and Entropy (ΔS°) are determined from the van't Hoff equation: ( \ln Kc = -\frac{\Delta H^\circ}{R}\frac{1}{T} + \frac{\Delta S^\circ}{R} ). A plot of ( \ln Kc ) versus ( 1/T ) gives a straight line where the slope is ( -\Delta H^\circ/R ) and the intercept is ( \Delta S^\circ/R ) [91].
Kinetic Studies for Activation Energy (Ea)
Objective: To determine the rate of adsorption and the activation energy (Ea), which quantifies the energy barrier of the process [90] [95].
Materials: Similar to those used in batch adsorption studies.
Procedure:
- Time-Dependent Adsorption: In a series of flasks, combine a fixed dose of adsorbent with a fixed volume of adsorbate solution at a known initial concentration and pH.
- Sampling: Agitate the flasks and collect samples at predetermined time intervals (e.g., 5, 10, 20, 30, 60, 120 minutes).
- Analysis: Immediately separate the adsorbent from each sample and analyze the adsorbate concentration in the supernatant.
Kinetic and Activation Energy Analysis:
- Fit the time-dependent adsorption data (( q_t ) vs. ( t )) to kinetic models such as the pseudo-first-order or pseudo-second-order model. The pseudo-second-order model has been frequently reported as the best fit for adsorption on porous carbons [92] [95].
- The activation energy (Ea) can be determined using the Arrhenius equation by conducting kinetic experiments at different temperatures and relating the rate constant (( k )) to temperature: ( \ln k = \ln A - \frac{Ea}{RT} ), where ( A ) is the Arrhenius factor. A plot of ( \ln k ) versus ( 1/T ) yields a straight line with a slope of ( -Ea/R ) [95].
Advanced In-Situ Characterization
Objective: To directly observe and quantify ion adsorption and electrosorption mechanisms under operating conditions, providing deeper insight into thermodynamic drivers [10] [96].
Techniques:
- In-Situ Nuclear Magnetic Resonance (NMR) Spectroscopy: Used to directly observe and quantify aqueous adsorbate partitioning behavior driven by spontaneous physisorption within micropores. It can distinguish between ionophilic and ionophobic systems and reveal charge-balancing mechanisms (counter-ion adsorption vs. co-ion expulsion) during electrosorption [10].
- In-Situ X-ray Transmission (XRT) Measurements: Employed to study ion concentration changes within the electrode pores during electrochemical operation. This technique can identify whether counter-ion adsorption, co-ion expulsion, or ion replacement is the dominant charge storage mechanism and how it depends on salt concentration and charging velocity [96].
Visualization of Experimental Workflows and Relationships
Thermodynamic Validation Workflow
Ion Adsorption Pathway Energetics
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 4: Key Research Reagents and Materials for Ion Adsorption Studies
| Item | Function & Purpose | Example from Research |
|---|---|---|
| Activated Carbon (AC) | Primary adsorbent; high surface area and porosity enable high adsorption capacity. | Derived from chickpea husks, green tea residues, hard wood composite, winemaking waste [92] [94] [91]. |
| Chemical Activating Agents | Used in synthesis to develop high porosity in the carbon structure. | KOH, K₂CO₃, H₃PO₄ [92] [94] [91]. |
| Model Adsorbates | Represent target pollutants or ionic species for standardized testing. | Methylene Blue (cationic dye), Methyl Red (anionic dye), Pb(II), Cu(II), Cd(II), Cr(VI) ions [92] [91]. |
| Aqueous Electrolytes | Medium for ion adsorption studies; composition affects solvation and mechanism. | Sodium sulfate, sodium bis(trifluoromethane)sulfonimide, RbBr, CsCl, NaCl [10] [96]. |
| Spectrophotometers / AAS | Quantify adsorbate concentration before and after adsorption. | UV-Vis Spectrophotometer (for dyes), Atomic Absorption Spectrometer (for metals) [92] [91] [95]. |
| Surface Characterization Tools | Analyze adsorbent's physical and chemical properties. | BET Surface Area Analyzer, SEM, FTIR, Boehm Titration [92] [94] [91]. |
| In-Situ Probes | Reveal real-time ion behavior and mechanisms during adsorption/electrosorption. | NMR Spectroscopy, X-ray Transmission (XRT) [10] [96]. |
Long-Term Performance and Regeneration Potential of Porous Carbon Electrodes
Porous carbon (PC) electrodes are central to advancing sustainable electrochemical technologies, from capacitive deionization (CDI) for water desalination to energy storage in supercapacitors. Their long-term performance and regeneration potential are critical factors determining economic viability and environmental sustainability. This whitepaper examines the fundamental mechanisms governing performance degradation and explores advanced regeneration strategies that restore electrode capacity. Within the broader thesis of ion adsorption research, we analyze how pore architecture, surface chemistry, and operational parameters influence ion trapping and release kinetics. The findings presented herein provide researchers with a technical framework for developing durable, high-performance carbon electrodes capable of maintaining operational efficiency over extended lifecycle periods.
The long-term performance of porous carbon electrodes is intrinsically linked to their ion adsorption capabilities, a process governed by complex interfacial phenomena. Capacitive deionization and supercapacitors primarily store energy or remove ions via electric double-layer (EDL) formation, where ions from the electrolyte are electrostatically adsorbed onto the pore surfaces of the carbon electrode [9] [74]. However, during extended cycling, especially under high-power conditions, a gradual but inevitable degradation of performance occurs, manifesting as reduced salt adsorption capacity in CDI or diminished specific capacitance in energy storage devices [97].
A primary mechanism of performance degradation is ion trapping, where certain ions become irreversibly confined within the complex pore network of the carbon electrode and cannot be released during the reverse charging step. This phenomenon is particularly pronounced under high-current-density operation [97]. The confinement is not solely a physical spatial restriction; in cationic porous organic frameworks, an "ionic framework-induced secondary confinement" occurs, where electrostatic interactions between the working ions (e.g., Li⁺, PF₆⁻) and the native ions of the framework (e.g., TFSI⁻) lead to the formation of aggregated ion pairs that are kinetically hindered from escaping [97]. Advanced characterization techniques, including in situ Raman spectroscopy, have directly identified distinctive signals of aggregated TFSI⁻ anions during high-rate cycling, confirming this ion pairing effect [97].
Furthermore, the ion desolvation state significantly influences adsorption thermodynamics and kinetics. Within the nanoconfined pores of carbon, potassium ions can exist in five distinct desolvation states, denoted as [K(H₂O)₀₋₄]⁺, each with its own desolvation energy and diffusion barrier [15]. An imbalance in these energies can lead to preferential stabilization of certain ion states, contributing to their trapping and a consequent decline in accessible adsorption sites over time. Understanding these fundamental adsorption and confinement mechanisms is the first step toward developing effective regeneration protocols to reverse capacity fade and extend electrode service life.
Performance Characterization and Quantitative Data
The long-term efficacy of porous carbon electrodes is quantified through standardized electrochemical and desalination metrics. The tables below consolidate key performance indicators and the impact of various degradation mitigation strategies based on recent research.
Table 1: Key Performance Metrics for Porous Carbon Electrodes
| Performance Metric | Definition | Significance for Long-Term Performance | Typical Range/Value |
|---|---|---|---|
| Cycle Life | Number of charge/discharge cycles before capacity falls below 80% of initial value. | Direct measure of electrode durability and longevity. | Varies widely; up to 60,000 cycles demonstrated with refresh strategies [97]. |
| Specific Capacitance (F/g) | Charge stored per unit mass of electrode material. | Indicator of the energy storage or ion adsorption capacity. | Up to 273 F g⁻¹ reported for optimized, coal-derived PC [15]. |
| Capacity Retention | Percentage of initial capacity retained after a specified number of cycles. | Quantifies the rate of performance degradation over time. | Can drop to ~72% after 10,000 cycles at high rate; refreshable to ~97% [97]. |
| Coulombic Efficiency | Ratio of discharge to charge capacity in a cycle. | Reflects charge reversibility; losses indicate parasitic reactions or ion trapping. | Decreases with cycling if irreversible side reactions occur. |
Table 2: Strategies for Enhancing Long-Term Performance and Regeneration
| Strategy | Mechanism of Action | Impact on Longevity & Regeneration | Key Findings |
|---|---|---|---|
| Capacity Refreshing [97] | Intermittent low-current cycling to release trapped ions from the porous framework. | Extends cycle life significantly by reactivating trapped charge carriers. | Restored capacity from 110 mAh/g to 148 mAh/g after 10,000 cycles; enabled 60,000 cycles at 20 C. |
| Surface Functionalization [31] | Coating PC with metal oxides (e.g., Mn₂O₃) or metals (e.g., Co) to modify surface charge and ion affinity. | Improves electrode kinetics and reduces charge transfer resistance, mitigating degradation. | Functionalized electrodes showed larger potential window, improved rate capability, and lower Li⁺ transfer resistance. |
| Entropy-Driven Design [13] | Creating "high-entropy carbon" with distorted carbon rings and multi-element doping to provide diverse ion adsorption sites. | Enhances intrinsic capacitance and stability through a thermodynamically stabilized disordered structure. | Proposed novel capacitance storage mechanisms via unit, ring, and element entropy. |
| Defect Engineering [9] | Introducing topological defects (e.g., five-/seven-membered carbon rings) and heteroatoms (O, N, S) to tune electronic properties. | Increases active sites for ion adsorption and can improve hydrophilicity and conductivity. | Optimized surface chemistry synergizes with specific ion desolvation states to enhance EDL capacitance [15]. |
Regeneration Mechanisms and Methodologies
The Capacity Refresh Protocol
A seminal study demonstrated a highly effective capacity refreshing strategy for a porous organic framework electrode [97]. The protocol is designed to counteract the capacity fade experienced during high-power cycling.
Experimental Workflow:
- High-Rate Cycling: The electrode is subjected to extended cycling (e.g., 10,000 cycles) at a high current rate (20 C, or 6 A/g), leading to a measurable drop in discharge capacity (e.g., from 153 mAh/g to 110 mAh/g).
- Refresh Phase: A short series of charge/discharge cycles (as few as 10 cycles) is applied at a significantly lower current rate (0.5 C).
- Performance Validation: The electrode is returned to the high-rate (20 C) operation, where its capacity is measured and shown to be restored to a level close to its initial value (148 mAh/g).
This refresh cycle can be repeated periodically, enabling the electrode to withstand over 60,000 cycles at a high rate, a lifespan that surpasses most conventional organic electrodes.
Underlying Mechanism: The refresh mechanism is rooted in reversing "ionic framework-induced secondary confinement" [97]. During high-rate cycling, ions do not have sufficient time to fully dissociate and diffuse out of the framework channels. Instead, they form kinetically trapped aggregates, such as Li⁺-TFSI⁻ pairs. The application of a low current rate during the refresh phase provides a milder thermodynamic driving force and a longer timescale, allowing these trapped ion pairs to gradually dissociate and be released from the framework, thereby reactivating the adsorption sites. In situ Fourier transform infrared (FTIR) and Raman spectroscopy confirmed the recovery of triazine segment signals and the weakening of aggregated TFSI⁻ signals after the refresh process [97].
Ion Desolvation State Management
The regeneration potential is also influenced by the fundamental thermodynamics of ion adsorption. Research has shown that potassium ions within realistic porous carbons exist in a distribution of five distinct desolvation states, [K(H₂O)₀₋₄]⁺, each with a characteristic desolvation energy and diffusion barrier [15]. Managing these states is key to optimizing capacitance and facilitating regeneration. A comprehensive design strategy based on a dual thermodynamic-kinetic optimization principle has been proposed to enhance the electric double-layer capacitance. This involves identifying the appropriate types and concentrations of surface oxygen groups that synergize with specific ion desolvation states, thereby creating a more favorable energy landscape for both ion adsorption and desorption during cycling [15].
Experimental Protocols for Performance and Regeneration Analysis
This section outlines detailed methodologies for evaluating the long-term performance and regeneration potential of porous carbon electrodes, focusing on electrochemical cycling and material characterization.
Protocol: Electrochemical Cycling and Capacity Refresh Test
Objective: To determine the cycle life of a porous carbon electrode under high-power conditions and evaluate the efficacy of a intermittent low-current refresh protocol in restoring lost capacity [97].
Materials:
- Porous Carbon Electrode: Fabricated into a working electrode.
- Counter Electrode: Lithium metal foil.
- Electrolyte: 1 M LiPF₆ in a suitable organic solvent.
- Equipment: Electrochemical workstation (e.g., Biologic VMP-3) or battery cycler (e.g., Arbin BT2000), glove box filled with argon.
Procedure:
- Cell Assembly: Assemble a coin cell (e.g., CR2032) or a multi-electrode swagelok cell inside an argon-filled glove box, with the PC electrode as the working electrode and Li metal as the counter/reference electrode, separated by a glass fiber separator soaked with electrolyte.
- Initial Characterization: Perform cyclic voltammetry (e.g., 0.1 to 3.0 V vs. Li/Li⁺ at 0.5 mV/s) and electrochemical impedance spectroscopy (EIS, e.g., 100 kHz to 10 mHz) to characterize the initial state of the electrode.
- Baseline Capacity Measurement: Conduct 5 galvanostatic charge-discharge (GCD) cycles at a low current rate (e.g., 0.2 C) to determine the initial stable discharge capacity of the electrode.
- High-Rate Aging: Subject the cell to continuous GCD cycles at a high current rate (e.g., 20 C, 6 A/g). Record the discharge capacity every 100 cycles.
- Implement Refresh Cycle: After a predetermined number of cycles (e.g., 10,000) or when capacity fade exceeds a set threshold (e.g., 20%), interrupt the high-rate cycling. Apply a limited number of GCD cycles (e.g., 10 cycles) at a low current rate (e.g., 0.5 C).
- Post-Refresh Performance: Immediately resume GCD cycling at the high rate (20 C) and measure the recovered discharge capacity.
- Repetition: Repeat steps 4-6 to demonstrate the periodicity and robustness of the refresh protocol over a total of tens of thousands of cycles.
Data Analysis:
- Plot discharge capacity versus cycle number to visualize the degradation profile and the recovery peaks after each refresh intervention.
- Calculate capacity retention before and after each refresh cycle.
- Fit EIS data to an equivalent circuit model to track changes in charge-transfer resistance (Rₑₜ) throughout the aging and refresh process.
Protocol: Characterization of Ion Trapping and Release
Objective: To directly observe the chemical state of the porous carbon framework and the trapped ions before and after the refresh process using in situ spectroscopic techniques [97].
Materials:
- Porous Carbon Electrode
- Specialized Equipment: In situ electrochemical cell compatible with FTIR and Raman spectrometers.
Procedure:
- Setup: Place the assembled in situ electrochemical cell in the spectrometer.
- Initial Spectrum: Acquire FTIR and Raman spectra of the pristine electrode at the open-circuit potential.
- Spectra During Aging: During the high-rate cycling (Step 4 of Protocol 4.1), periodically pause at a specific state-of-charge (e.g., fully charged) and acquire FTIR and Raman spectra.
- Spectra After Aging: After significant capacity fade, acquire spectra at the same state-of-charge as in step 3.
- Spectra During/After Refresh: Acquire spectra during or immediately after the application of the low-current refresh cycles (Step 5 of Protocol 4.1).
- Data Interpretation:
- FTIR: Monitor the characteristic peaks of specific functional groups in the carbon framework (e.g., triazine rings at ~1605 and 1504 cm⁻¹). A weakening of these signals indicates ion trapping, while their recovery after refresh indicates ion release [97].
- Raman: Monitor peaks associated with aggregated ions (e.g., aggregated TFSI⁻ at ~754 cm⁻¹) and framework redox states (e.g., bipyridine radical cation V•+ at ~1647 cm⁻¹). A decrease in aggregation-related peaks after refresh confirms the dissolution of ion pairs [97].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Reagents and Materials for Research on PC Electrodes
| Item Name | Function/Application | Technical Notes & Examples |
|---|---|---|
| Green Activators [9] | Sustainable alternatives to traditional corrosive chemicals (KOH, HF) for creating porosity in carbon precursors. | Includes green inorganic salts (e.g., NaCl, Na₂CO₃) and organic salts (e.g., citrate, gluconate). Reduces environmental impact and equipment corrosion. |
| Hard & Soft Templates [74] | To create precisely controlled pore sizes and hierarchical pore structures during carbon synthesis. | Hard templates: SiO₂, MgO, ZnO nanoparticles. Soft templates: block copolymers (e.g., Pluronic F127). |
| Heteroatom Dopants [9] [13] | To modify the electronic structure and surface chemistry of carbon, enhancing conductivity and introducing pseudocapacitance. | Common precursors: Urea (for N-doping), Thiourea (for S,N-doping), Boric acid (for B-doping), Phytic acid (for P-doping). |
| Metal Oxide Nanoflowers [31] | As hierarchical void-fillers or thin-film coatings on PC to enhance lithium-ion storage kinetics and capacity. | Examples: MnCo₂O₄, TiO₂. Provide abundant active sites and lower charge-transfer resistance. |
| Lithium Salts & Organic Electrolytes [31] [97] | As the electrolyte system for evaluating performance in lithium-ion capacitors or battery configurations. | Common salt: LiPF₆. Solvents: Ethylene Carbonate (EC), Diethyl Carbonate (DEC) mixtures. Handling requires an inert atmosphere glove box. |
| In Situ Electrochemical Cells [97] | To allow simultaneous electrochemical cycling and spectroscopic characterization (Raman, FTIR, UV-Vis) of the electrode material. | Crucial for real-time monitoring of ion trapping/refresh mechanisms and structural changes in the working electrode. |
Visualization of Degradation and Refresh Pathways
Understanding the molecular-level interactions that lead to degradation and enable regeneration is crucial for material design. The following diagram synthesizes the key pathways and mechanisms involved in the ion trapping and refresh process, as revealed by recent studies.
Conclusion
The study of ion adsorption in porous carbon electrodes reveals a rapidly advancing field where material design is becoming increasingly sophisticated. The integration of entropy-driven concepts, such as high-entropy carbon with tailored defects and multi-element doping, provides a novel pathway for creating materials with superior ion uptake and selectivity. Methodological advances in using sustainable biomass precursors and precise activation protocols enable the cost-effective production of high-performance carbons. The optimization of parameters like pH, pore structure, and surface chemistry is paramount for achieving specific application goals, whether for high-power supercapacitors or the targeted removal of toxic heavy metals. Validation through advanced in situ techniques and robust comparative analysis confirms that these materials are not only effective but also predictable in their behavior. Future directions should focus on translating these material platforms into biomedical devices, such as detoxification systems or drug delivery platforms, leveraging their high surface area and tunable adsorption properties for clinical applications. The convergence of materials science, electrochemistry, and environmental engineering outlined here paves the way for innovative solutions in both energy and health.
References
- 1. - Definition, Adsorption and Types - GeeksforGeeks Mechanism [https://www.geeksforgeeks.org/chemistry/adsorption-definition-mechanism-and-types/]
- 2. byjus.com/jee/ adsorption [https://byjus.com/jee/adsorption/]
- 3. Entropy-driven disordered porous carbon ... - RSC Publishing [https://pubs.rsc.org/en/content/articlehtml/2025/cc/d5cc05329c]
- 4. High-Entropy Materials: A New Paradigm in the Design of ... [https://link.springer.com/article/10.1007/s40820-025-01842-w]
- 5. sciencedirect.com/topics/chemistry/ adsorption - entropy [https://www.sciencedirect.com/topics/chemistry/adsorption-entropy]
- 6. (PDF) The Entropies of Adsorbed Molecules [https://www.academia.edu/2404149/The_Entropies_of_Adsorbed_Molecules]
- 7. Recent advances in the high entropy materials for ... [https://www.sciencedirect.com/science/article/pii/S2666935825000679]
- 8. Carbon/High-Entropy Alloy Nanocomposites: Synergistic ... [https://www.mdpi.com/2313-0105/11/9/317]
- 9. Engineering sustainable porous carbon electrodes for ... [https://www.sciencedirect.com/science/article/abs/pii/S0010854525005193]
- 10. Solvation effects on aqueous ion adsorption and ... [https://www.sciencedirect.com/science/article/pii/S0008622324007504]
- 11. Nuclear Magnetic Resonance Studies of Aqueous Electrolyte ... [https://eprints.lancs.ac.uk/id/eprint/228213/]
- 12. Quantitative and qualitative analysis of ionic solvation ... [https://pubs.rsc.org/en/content/articlelanding/2014/ra/c3ra48051h]
- 13. Entropy-driven disordered porous carbon ... - RSC Publishing [https://pubs.rsc.org/en/content/articlehtml/2025/cc/d5cc05329c?page=search]
- 14. Biostructure-Inspired High Entropy Carbon-Like Material ... [https://www.sciencedirect.com/science/article/pii/S1359836825009965]
- 15. Decoding Potassium Ion Desolvation States for Enhanced ... [https://pubmed.ncbi.nlm.nih.gov/41243658/]
- 16. About CDI - CDI & electrosorption [https://www.cdi-electrosorption.org/sample-page/]
- 17. Modulating Pore Channels of Activated Carbon from ... [https://www.sciencedirect.com/science/article/pii/S236996982500057X]
- 18. Nitrogen and oxygen-codoped dense porous carbon ... [https://www.oaepublish.com/articles/energymater.2025.105]
- 19. Electrosorption at metal surfaces from first principles [https://www.nature.com/articles/s41524-020-00394-4]
- 20. Ionic Liquid Derived Heteroatom-adaptive Porous Carbon ... [https://www.sciencedirect.com/science/article/abs/pii/S0008622325006955]
- 21. Understanding the role of confinement in the behavior ... [https://www.sciencedirect.com/science/article/abs/pii/S0167732224027958]
- 22. Mesoscale dynamics of electrosorbed ions in fast-charging ... [https://pubmed.ncbi.nlm.nih.gov/40550973/]
- 23. Advanced carbon electrodes for supercapacitors [https://www.sciexplor.com/articles/smd.2025.0008]
- 24. Current Trends in Synthesis and Characterization of ... [https://www.mdpi.com/2673-8783/5/4/70]
- 25. Recent progress in the development of biomass-derived ... [https://pubs.rsc.org/en/content/articlelanding/2021/ta/d0ta09706c]
- 26. Biomass Derived N-Doped Porous Carbon Made from ... [https://www.mdpi.com/1420-3049/28/12/4633]
- 27. Advanced nitrogen-doped wood-derived biocarbon for ... [https://www.sciencedirect.com/science/article/pii/S0169433225021221]
- 28. Solanaceous Crops-Derived Nitrogen-Doped Biomass ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12430359/]
- 29. Nitrogen-doped hierarchical porous carbons derived from ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10311552/]
- 30. Understanding Sorption of Aqueous Electrolytes in Porous ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11009947/]
- 31. Lithium-ion adsorption on surface modified porous carbon [https://www.sciencedirect.com/science/article/abs/pii/S2352152X23016183]
- 32. Hierarchical pore engineering of lignocellulose-based ... [https://www.sciopen.com/article/10.26599/NR.2025.94907960]
- 33. Thermal tuning of nanocomposites for superior cadmium ... [https://www.nature.com/articles/s41598-025-09274-7]
- 34. Hemp-Derived Hierarchical Porous Carbon with an ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12612924/]
- 35. Transforming waste biomass into oxygen-rich hierarchical ... [https://www.sciencedirect.com/science/article/abs/pii/S0378775325026485]
- 36. Wrinkled hierarchical porous carbon spheres with ... [https://www.nature.com/articles/s41545-024-00372-z]
- 37. BET Theory [https://wiki.anton-paar.com/en/bet-theory/]
- 38. How reproducible are surface areas calculated from the ... [https://www.nist.gov/publications/how-reproducible-are-surface-areas-calculated-bet-equation]
- 39. Structural descriptors controlling pore-filling mechanism in ... [https://pubs.rsc.org/en/content/articlelanding/2025/eb/d5eb00210a]
- 40. Application of fourier transform infrared (FTIR) ... [https://www.sciencedirect.com/science/article/pii/S2772582024000214]
- 41. FT-IR Spectroscopy Provides Insights into Green Synthesis ... [https://www.spectroscopyonline.com/view/ft-ir-spectroscopy-provides-insights-into-green-synthesis-and-stabilization-of-nanoparticles]
- 42. The role of biomass elemental composition and ion ... [https://www.sciencedirect.com/science/article/pii/S0045653522041686]
- 43. An in situ SEM-FIB-based method for contrast ... [https://pubmed.ncbi.nlm.nih.gov/25088604/]
- 44. Integrating experimental and theoretical investigations of ... [https://www.sciencedirect.com/science/article/pii/S2590123024011484]
- 45. The principle of BET method | Universal Lab Blog [https://universallab.org/blog/BET-method-introduction]
- 46. Adsorptive Removal of Lead Ions from Wastewater Using ... [https://www.sciencedirect.com/science/article/pii/S2772416625003110]
- 47. Batch experimental studies on Acid Blue 25 dye removal ... [https://www.nature.com/articles/s41598-025-17781-w]
- 48. Incorporation of transition metal-oxide on 3D porous ... [https://www.sciencedirect.com/science/article/abs/pii/S001346862500698X]
- 49. Cu-Doped Porous Carbon Derived from Heavy Metal ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6630645/]
- 50. Adsorption of heavy metals from water using teak-based ... [https://www.sciencedirect.com/science/article/pii/S2588913325000304]
- 51. Hierarchical Porous Carbon Electrodes with Sponge-Like ... [https://www.mdpi.com/1424-8220/21/4/1346]
- 52. Isotherm models for adsorption of heavy metals from water [https://www.sciencedirect.com/science/article/abs/pii/S0045653522020380]
- 53. Isotherm models for adsorption of heavy metals from water [https://pubmed.ncbi.nlm.nih.gov/35787879/]
- 54. Optimization of porous carbon structure through high ... [https://www.sciencedirect.com/science/article/abs/pii/S0013468625010837]
- 55. Influence of Pyrolysis Temperature on the Properties and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12383750/]
- 56. Redefining the roles of alkali activators for porous carbon [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d4sc07145j]
- 57. Adsorption of Phenol from Aqueous Solution using ... [https://link.springer.com/article/10.1007/s11270-025-08431-y]
- 58. Effect of hydrothermal pH values on the morphology ... [https://www.sciencedirect.com/science/article/abs/pii/S0960852423015997]
- 59. Electrosorption of alkaline earth metal ions onto activated ... [https://www.sciencedirect.com/science/article/abs/pii/S0011916424009834]
- 60. Thermodynamic, isotherm and kinetic studies lead ions ... [https://www.nature.com/articles/s41598-025-09307-1]
- 61. The Adsorption Performance of Porous Activated Carbons ... [https://www.mdpi.com/2312-7481/9/6/151]
- 62. Implications of apparent pseudo-second-order adsorption ... [https://bioresources.cnr.ncsu.edu/resources/implications-of-apparent-pseudo-second-order-adsorption-kinetics-onto-cellulosic-materials-a-review/]
- 63. Kinetic and mechanistic study of CO2 adsorption on ... [https://www.sciencedirect.com/science/article/pii/S2212982024000519]
- 64. Preparation and Application of Electrodes in Capacitive ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4740477/]
- 65. Understanding adsorption mechanisms and metal ion ... [https://www.sciencedirect.com/science/article/pii/S1387181124001811]
- 66. Langmuir Isotherm Equation - an overview [https://www.sciencedirect.com/topics/engineering/langmuir-isotherm-equation]
- 67. Freundlich Equation - an overview [https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/freundlich-equation]
- 68. Freundlich Isotherm: An Adsorption Model Complete ... [https://www.mdpi.com/2076-3417/11/17/8078]
- 69. Adsorption, Equilibrium Isotherm, and Thermodynamic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8537514/]
- 70. Enhancement of electrode surface hydrophilicity and ... [https://www.sciencedirect.com/science/article/abs/pii/S0003267024012248]
- 71. Pore-structure control of porous carbon electrode materials ... [https://www.sciencedirect.com/science/article/abs/pii/S1572665724002091]
- 72. Understanding the effect of nanosized carbon texture on ... [https://www.sciencedirect.com/science/article/pii/S2405829725001722]
- 73. Research advances in carbon-based electrode materials ... [https://www.frontiersin.org/journals/materials/articles/10.3389/fmats.2025.1639589/full]
- 74. Recent Advances in Porous Carbon Materials as ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10254524/]
- 75. Adsorption of heavy metal onto biomass-derived activated ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9891085/]
- 76. Adsorptive removal of gaseous contaminants using biomass ... [https://pubs.rsc.org/sv/content/articlehtml/2025/ra/d4ra08572h]
- 77. Fabrication and characterization of coconut shell activated ... [https://www.sciencedirect.com/science/article/pii/S2211715622000108]
- 78. Fabrication of Bamboo-Based Activated Carbon for Low- ... [https://www.mdpi.com/2071-1050/16/4/1634]
- 79. Biomass-derived porous carbon highly efficient for removal ... [https://www.sciencedirect.com/science/article/pii/S2468025719300184]
- 80. Adsorption of Sulfonamides in Aqueous Solution on Reusable ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8813774/]
- 81. A comprehensive review of enhanced CO2 capture using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11409178/]
- 82. Mesoscopic simulations of the in situ NMR spectra ... [https://pubs.rsc.org/en/content/articlelanding/2021/cp/d1cp02130c]
- 83. Applications of nuclear magnetic resonance in exploring ... [https://www.sciencedirect.com/science/article/pii/S2772516224000792]
- 84. Unexpected Oversolubility of CO2 Measured at Electrode ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12512178/]
- 85. Assessing CO2 Capture in Porous Sorbents via Solid-State ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10141401/]
- 86. Quantifying the adsorption of flowing gas mixtures in ... [https://www.sciencedirect.com/science/article/abs/pii/S1387181117303670]
- 87. Revealing the “double-layer ionic monotonic adsorption ... [https://www.sciencedirect.com/science/article/abs/pii/S1385894725066379]
- 88. Multidimensional soft-hard heterostructure porous carbon ... [https://www.sciencedirect.com/science/article/abs/pii/S138589472510209X]
- 89. Activated carbon cloth as efficient microporous electrode for ... [https://pubs.rsc.org/en/content/articlehtml/2025/ma/d5ma00422e]
- 90. Unraveling the Ion Adsorption Kinetics in Microporous ... [https://pubmed.ncbi.nlm.nih.gov/33979129/]
- 91. Chemically activated carbon production from agricultural ... [https://link.springer.com/article/10.1007/s13201-019-0942-8]
- 92. Adsorption of Organic Compounds on Adsorbents ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9415288/]
- 93. The role of impurities in porous carbons for bioinspired ... [https://www.sciencedirect.com/science/article/pii/S1385894723056292]
- 94. Porous carbons prepared from a novel hard wood composite ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10687519/]
- 95. Removal of copper ions from wastewater by adsorption onto a ... [https://bioresources.cnr.ncsu.edu/resources/removal-of-copper-ions-from-wastewater-by-adsorption-onto-a-green-adsorbent-from-winemaking-wastes/]
- 96. Salt concentration and charging velocity determine ion ... [https://www.nature.com/articles/s41467-018-06612-4]
- 97. Refresh organic electrodes for high-power and long-cycle ... [https://www.nature.com/articles/s41467-025-60355-7]