Cyclic Voltammetry for Redox Reaction Analysis: A Comprehensive Guide from Fundamentals to Biomedical Applications
This article provides a comprehensive guide to Cyclic Voltammetry (CV) for the analysis of redox reactions, tailored for researchers, scientists, and drug development professionals.
Cyclic Voltammetry for Redox Reaction Analysis: A Comprehensive Guide from Fundamentals to Biomedical Applications
Abstract
This article provides a comprehensive guide to Cyclic Voltammetry (CV) for the analysis of redox reactions, tailored for researchers, scientists, and drug development professionals. It covers foundational principles, including the interpretation of voltammograms and key equations like Randles-Sevcik. The guide details methodological setup, from electrode selection to experimental parameters, and highlights diverse applications from neurotransmitter detection to anticancer drug analysis. It further offers practical troubleshooting advice for common experimental issues and explores advanced validation techniques, including the comparison with other analytical methods and the integration of computational models like Density Functional Theory (DFT) for predictive analysis.
Understanding Cyclic Voltammetry: Core Principles and Redox Theory
In the field of electrochemical research, the three-electrode system is a foundational setup for conducting precise and controlled experiments. It has become standard equipment in research laboratories for investigating reaction mechanisms, optimizing battery performance, and developing next-generation energy materials [1]. Unlike the two-terminal batteries used in daily life, the three-electrode system is a specialized laboratory configuration designed for accurate measurement and control [2].
This system's development was a significant advancement over the simpler two-electrode setup. Historically, two-electrode systems were used but had major drawbacks, particularly in measuring and controlling electrode potentials, which led to considerable errors. The introduction of the reference electrode in the 1920s, thereby creating the three-electrode system, greatly improved the precision and reproducibility of electrochemical experiments [2]. The core principle of this system is the separation of current control from potential measurement, enabling independent control of the working electrode potential while the counter electrode carries the system current [2].
The Roles and Requirements of the Three Electrodes
A typical three-electrode electrochemical cell consists of three distinct components: the Working Electrode (WE), the Reference Electrode (RE), and the Counter Electrode (CE), also known as the auxiliary electrode [2] [1]. Each plays a unique and critical role.
Working Electrode (WE)
The Working Electrode is the core of the electrochemical cell, where the reaction of interest occurs [2] [1]. Its properties are critical for obtaining meaningful data.
- Role: The WE is the electrode under study; the electrochemical reaction being investigated happens at its surface [2] [3].
- Requirements: An ideal working electrode should be chemically inert to the electrolyte, have a reproducible and uniform surface, and present a controlled geometric area [2] [1]. Its surface must be free of impurities, which often necessitates pre-treatment steps like polishing and cleaning before experiments [1] [3].
- Common Materials: Glassy carbon, platinum (Pt), gold (Au), silver (Ag), and conductive oxides such as FTO and ITO [2] [1].
Reference Electrode (RE)
The Reference Electrode acts as a stable potential benchmark in the system [1].
- Role: It provides a known, stable reference potential against which the working electrode's potential is measured and controlled [2]. Crucially, it is designed to carry virtually no current, ensuring its potential remains constant [2] [3].
- Requirements: An optimal reference electrode has a well-defined and stable potential, high exchange current density (making it non-polarizable), and does not react with the electrolyte [1]. It should also have a low temperature coefficient to minimize potential shifts with temperature changes [1].
- Common Materials: Silver/Silver Chloride (Ag/AgCl), Saturated Calomel Electrode (SCE), and Mercury/Mercuric Oxide (Hg/HgO) [1] [4]. For non-aqueous electrochemistry, pseudo-reference electrodes like a silver wire are also used, with their potential calibrated against an internal standard like ferrocene [3].
Counter Electrode (CE)
The Counter Electrode completes the electrical circuit, enabling current flow.
- Role: Also called the auxiliary electrode, the CE completes the current path and supplies/balances the current flowing to or from the working electrode [2] [3].
- Requirements: The counter electrode must be highly conductive and chemically stable to avoid unwanted side reactions [1]. It is typically chosen with a larger surface area than the working electrode to ensure that the current density is low, preventing it from becoming polarized and limiting the current [1] [4].
- Common Materials: Platinum wire or mesh, and graphite rods or plates [2] [1].
The following diagram illustrates the functional relationships and current flow within a standard three-electrode system.
(caption) Three-Electrode System Configuration. The potentiostat controls the WE potential versus the stable RE, while current flows between WE and CE.
Why a Three-Electrode System is Essential for Precise Research
The primary advantage of the three-electrode system over a two-electrode configuration is its ability to provide precise and unambiguous data. This is critical for advanced research techniques like cyclic voltammetry (CV), electrochemical impedance spectroscopy (EIS), and intermittent titration techniques (GITT/PITT) [2].
Precise Potential Control
The introduction of the reference electrode allows for independent measurement and control of the working electrode's potential without being influenced by the current flowing in the system. This greatly enhances experimental precision, especially when studying the kinetics and mechanisms of electrochemical reactions [2].
Elimination of Interfering Factors
In a two-electrode setup, voltage drops from solution resistance (known as the IR drop) and polarization of the counter electrode can obscure the true potential at the working electrode. The three-electrode cell, with its stable reference, eliminates much of this ambiguity, allowing researchers to more clearly separate and analyze different components within the electrochemical system [2].
Practical Evidence of Superiority
Comparative studies, particularly in sensing applications, demonstrate the practical benefits of the three-electrode system. For instance, research on a paper-based electrochemical aptasensor for dengue virus detection showed that the three-electrode setup had a substantially higher and more sensitive current response (ranging from 55.53 µA to 322.21 µA) compared to the two-electrode system (0.85 µA to 4.54 µA). This represents a current amplification of approximately 50 times, making the three-electrode method a more viable option for highly sensitive diagnostics [5].
The Researcher's Toolkit: Electrode Selection and Materials
Selecting the appropriate electrodes depends on several factors, including research objectives, electrolyte type (acidic, neutral, or alkaline), desired potential window, and sensitivity requirements [1]. The table below summarizes common choices for each electrode.
Table 1: Guide to Electrode Selection and Common Materials
| Electrode Type | Role & Key Characteristics | Common Materials & Applications |
|---|---|---|
| Working Electrode (WE) | Role: Site of the reaction of interest.Key Characteristics: Chemically inert, reproducible surface, controlled geometric area [2] [1]. | Glassy Carbon: Versatile; wide potential window [1].Platinum (Pt) & Gold (Au): Excellent conductivity; for electrocatalysis [1].Conductive Oxides (FTO/ITO): Essential for photoelectrochemistry [1]. |
| Reference Electrode (RE) | Role: Provides stable potential reference.Key Characteristics: Non-polarizable, stable and reproducible potential, minimal current draw [2] [1]. | Ag/AgCl: Very common for aqueous systems [1] [4].Saturated Calomel (SCE): Traditional standard for aqueous solutions [1] [4].Ag/Ag+ (non-aqueous): For organic solvent electrolytes [4]. |
| Counter Electrode (CE) | Role: Completes the current circuit.Key Characteristics: High conductivity, chemical stability, large surface area [2] [1]. | Platinum Mesh/Gauze: Inert, high surface area, ideal for most systems [1] [3].Graphite Rods: Cost-effective, chemically stable for long-duration tests [1]. |
Experimental Protocol: Setting Up a Three-Electrode Cell for Cyclic Voltammetry
This protocol outlines the steps for assembling a standard three-electrode cell and conducting cyclic voltammetry (CV), a fundamental technique for studying redox reactions.
Materials and Equipment
- Electrochemical Workstation (Potentiostat): The central control and measurement unit.
- Electrochemical Cell: e.g., a 50 mL glass beaker or specialized cell jar.
- Electrodes: Working, reference, and counter electrodes, selected based on Table 1.
- Electrolyte Solution: A high-purity salt (e.g., KCl, Na₂SO₄) dissolved in a solvent (water or organic), typically at concentrations of 0.1 M to 1.0 M.
- Connecting Cables & Clips: Alligator clips or other connectors for secure contact.
- Polishing Supplies: For working electrode preparation, including alumina or diamond polishing powders and microcloth pads.
Step-by-Step Procedure
Working Electrode Preparation:
- Polishing: Polish the surface of the solid working electrode (e.g., glassy carbon) sequentially with progressively finer alumina slurries (e.g., 1.0 µm, 0.3 µm, 0.05 µm) on a microcloth pad [3]. A mirror-finish is the goal.
- Rinsing: Rinse the electrode thoroughly with deionized water after each polishing step and after the final polish to remove all abrasive particles [3].
- Drying: Gently dry the electrode surface with a clean, lint-free tissue (e.g., Kimwipe) [6].
Cell Assembly:
- Add Electrolyte: Fill the electrochemical cell with the prepared electrolyte solution.
- Position Electrodes: Immerse the three electrodes into the electrolyte.
- Secure Connections: Use the connecting cables and clips to link the electrodes to the potentiostat. The red (working drive) and orange (working sense) leads are connected to the WE, the white (reference sense) lead to the RE, and the green (counter drive) lead to the CE [7].
Instrument Configuration:
- Turn on the potentiostat and connected computer.
- In the control software, select the cyclic voltammetry technique.
- Set the initial potential, upper vertex potential, and lower vertex potential based on the redox system under investigation.
- Set the scan rate (e.g., 50 mV/s or 100 mV/s). Multiple cycles are often run to check for reproducibility [3].
Running the Experiment and Data Acquisition:
- Initiate the CV scan. The instrument will automatically cycle the potential between the set limits.
- The potentiostat will record the current response at the working electrode, generating a current-potential plot known as a cyclic voltammogram.
- Run several cycles to ensure the signal is stable and reproducible [3].
Post-Experiment Shutdown and Cleaning:
- Once the experiment is complete, stop the measurement.
- Disconnect the electrodes from the potentiostat before removing them from the solution.
- Clean the working electrode according to material specifications and store all electrodes properly. Reference electrodes should be stored in an appropriate solution (e.g., Ag/AgCl in KCl) to prevent the porous frit from drying out [3].
Critical Troubleshooting Tips
- Non-Reproducible CVs: If consecutive cycles are not identical, the working electrode surface may be changing. Re-polish the electrode to ensure a clean, reproducible surface [3].
- Noisy or Unstable Signal: Check all electrical connections for secure contact. Ensure the counter electrode has a sufficiently large surface area to prevent polarization [1].
- Drifting Potential: This can indicate a faulty or contaminated reference electrode. Check the reference electrode's integrity and replace or refurbish it if necessary [3].
Advanced Applications in Research and Development
The three-electrode system's precision makes it indispensable across various advanced research fields.
- Battery Material Development: It is crucial for analyzing the electrochemical properties of electrode materials, such as diffusion coefficients and redox reaction rates. Techniques like GITT and PITT are used to optimize material design and battery performance [2] [4].
- Drug Development and Precision Medicine: Recent research combines conductive polymers with 3D printing to create electro-active drug delivery systems (DDS). These DDS use a three-electrode setup to apply precise voltages, enabling programmable, on-demand drug release profiles, which opens new avenues for personalized therapies [8].
- Biosensing: As demonstrated with the dengue virus aptasensor, the three-electrode system provides the sensitivity required for low-concentration detection of biological targets, making it valuable for medical diagnostics [5].
The three-electrode system is a cornerstone of modern electrochemical research. By separating the functions of potential measurement and current control, it provides an unparalleled level of precision for investigating redox reactions, characterizing new materials, and developing advanced diagnostic and therapeutic technologies. Its continued use is fundamental to progress in fields ranging from energy storage to precision medicine.
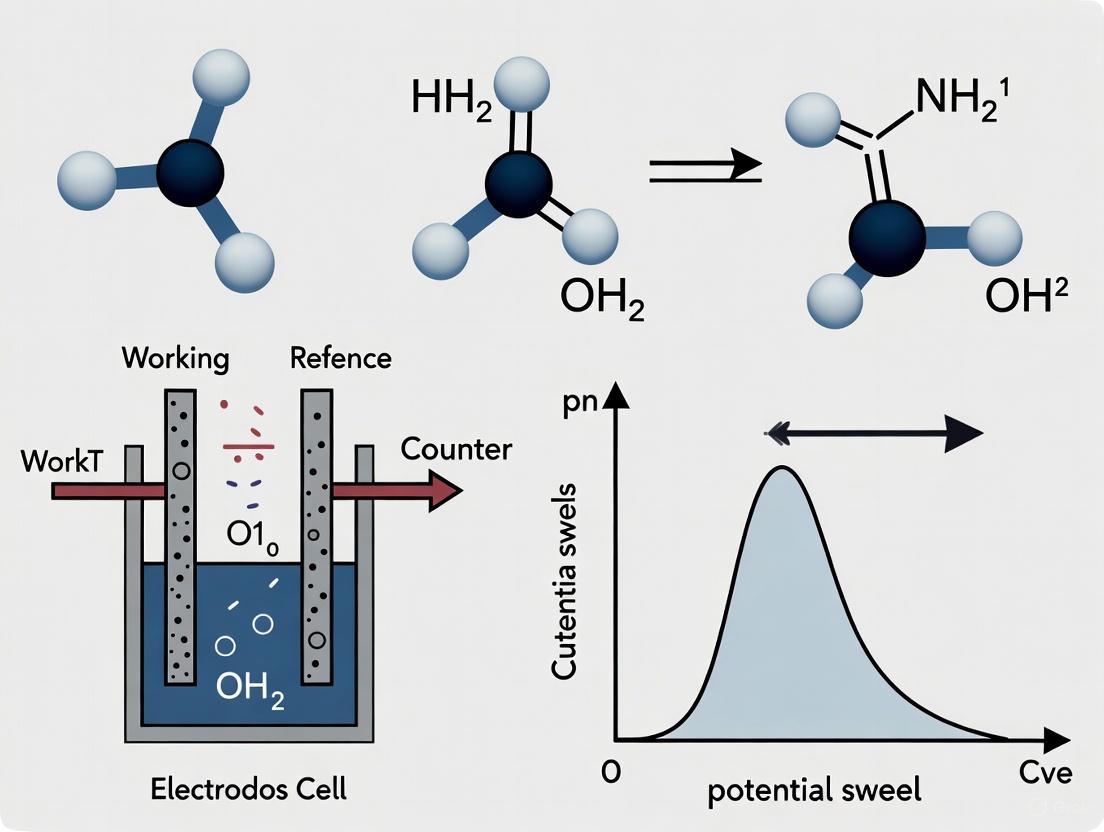
Cyclic voltammetry (CV) is a powerful and versatile electrochemical technique employed to rapidly elucidate information about the thermodynamics of redox processes, the energy levels of analytes, and the kinetics of electron-transfer reactions [9]. It is a fundamental method in the characterization of conductive polymers, battery materials, supercapacitors, fuel cell components, and pharmaceutical compounds [9] [10]. The technique involves measuring the current response of a redox-active solution to a linearly cycled potential sweep between the working and reference electrodes using a potentiostat [9]. The resulting plot of current versus potential often produces a characteristic "duck-shaped" profile, the cyclic voltammogram, which provides a wealth of qualitative and quantitative information to the trained researcher. This application note details the key features of this plot and outlines standardized protocols for its analysis within the context of redox reaction analysis research.
The Core Principles and Setup
The Three-Electrode System
A critical component of a reliable CV experiment is the three-electrode system, which separates the role of referencing the applied potential from the role of balancing the current produced [9].
- Working Electrode (WE): This is where the redox reaction of interest occurs. Its potential is varied linearly with time relative to the reference electrode. Common materials include glassy carbon, platinum, and gold.
- Reference Electrode (RE): This electrode maintains a fixed, well-known potential, providing a stable reference against which the working electrode's potential is controlled. Examples include Ag/AgCl and saturated calomel electrodes (SCE). It is designed to pass minimal current to avoid contamination and potential drift [9].
- Counter Electrode (CE): Also known as the auxiliary electrode, its primary function is to complete the electrical circuit by balancing the current generated at the working electrode. It typically has a larger surface area than the working electrode and is often made from an inert material like platinum wire [9].
The Potential Sweep and Redox Events
In a typical cyclic voltammetry experiment, the potentiostat sweeps the potential applied to the working electrode linearly over time. The scan starts at an initial potential, moves to a vertex potential, and then reverses direction to return to the initial potential [9]. During the forward sweep, if the potential is swept in a positive direction, an electroactive species may lose an electron in an oxidation (e.g., Fc → Fc⁺ + e⁻ for ferrocene). During the reverse sweep, the potential moves in a negative direction, and the oxidized species may gain an electron in a reduction (e.g., Fc⁺ + e⁻ → Fc) [9]. The current generated is a result of electron transfer between the redox species and the electrodes, and is carried through the solution by the diffusion and migration of ions, forming a capacitive electrical double layer at the electrode surface [9].
Deciphering the 'Duck-Shaped' Cyclic Voltammogram
The canonical "duck-shaped" voltammogram is the direct result of the processes described above. The current response is dependent on the concentration of the redox species at the working electrode surface, which is governed by diffusion [9]. The following walkthrough and diagram describe the formation of this shape.
Figure 1: The characteristic 'duck-shaped' cyclic voltammogram, showing the key points in the potential sweep and the corresponding electrochemical events at the working electrode [9].
- Point a: The initial potential is not sufficient to drive the oxidation or reduction of the analyte, resulting in negligible faradaic current [9].
- Point b: As the potential approaches the standard potential of the redox couple, the onset of oxidation (Eonset) is reached, and the current begins to increase exponentially [9].
- Point c: The current reaches a maximum, known as the anodic peak current (ipa), at the anodic peak potential (Epa). The current peak occurs because the depletion of the oxidant near the electrode surface (which decreases the current) begins to outweigh the effect of the increasing potential (which increases the current) [9].
- Point d: After the peak, the current decays as the system becomes limited by the mass transport of fresh analyte from the bulk solution to the electrode surface [9].
- Point e: Upon scan reversal, oxidation continues until the potential is sufficiently negative to reduce the oxidized species that have accumulated at the electrode surface [9].
- Point f: The cathodic peak current (ipc) is observed at the cathodic peak potential (Epc), corresponding to the reduction of the previously generated species [9].
Quantitative Data Analysis
The key quantitative parameters obtained from a cyclic voltammogram are the peak potentials (Ep) and the peak currents (ip), as illustrated in Figure 1 [11]. These parameters are used to determine the reversibility of a redox system and to calculate critical kinetic and thermodynamic properties.
Criteria for Reversibility
A redox system is considered electrochemically reversible if it remains in equilibrium throughout the potential scan, maintaining surface concentrations dictated by the Nernst equation [11]. The following criteria are used to assess reversibility:
Table 1: Diagnostic Criteria for a Reversible Redox Process in Cyclic Voltammetry [11].
| Parameter | Mathematical Relationship | Value at 25 °C |
|---|---|---|
| Peak Potential Separation | ΔEp = Epc - Epa | 59.2 / n mV |
| Peak Current Ratio | ipa / ipc | 1 |
| Scan Rate Dependence | ip / ν¹/² | Independent of scan rate (ν) |
The Randles-Sevcik Equation
For a reversible, diffusion-controlled process, the peak current (ip) is directly proportional to the concentration of the analyte and the square root of the scan rate. This relationship is described by the Randles-Sevcik equation [9] [11].
At 298 K, the equation is: ip = (2.69 × 10⁵) n³/² A C D¹/² ν¹/²
Table 2: Parameters of the Randles-Sevcik Equation.
| Symbol | Parameter | Typical Units |
|---|---|---|
| ip | Peak Current | Amperes (A) |
| n | Number of electrons transferred per molecule | Dimensionless |
| A | Electrode surface area | cm² |
| C | Bulk concentration of the analyte | mol cm⁻³ |
| D | Diffusion coefficient | cm² s⁻¹ |
| ν | Potential scan rate | V s⁻¹ |
This relationship allows researchers to determine the diffusion coefficient (D) of an analyte or verify the number of electrons (n) transferred in a redox process if the other parameters are known [9].
Irreversible and Quasi-Reversible Processes
Departures from the ideal reversible behavior occur for two major reasons:
- Slow Electron Transfer Kinetics: If the electron transfer kinetics (denoted by the standard heterogeneous electron transfer rate constant, kₛ) are too slow to maintain Nernstian equilibrium at the scan rate (ν) used, the process is termed quasi-reversible. This is characterized by a peak potential separation (ΔEp) greater than 59.2/n mV, with the value increasing with increasing scan rate [11].
- Chemical Reactions of O and R: If the oxidized (O) or reduced (R) species undergoes a following chemical reaction, the voltammogram is distorted. This often manifests as a decrease in the peak current ratio (ipa/ipc < 1), as not all of the molecules reduced on the forward scan are available for reoxidation on the reverse scan [11].
Experimental Protocol: Basic CV Experiment
Research Reagent Solutions and Materials
Table 3: Essential Materials for a Cyclic Voltammetry Experiment.
| Item | Function | Example |
|---|---|---|
| Potentiostat | Instrument to control potential and measure current. | Ossila Potentiostat, commercial systems. |
| Electrochemical Cell | Container for the electrolyte and analyte solution. | Glass vial or specialized cell. |
| Working Electrode | Site of the redox reaction; its material can affect reaction kinetics. | Glassy Carbon, Platinum, Gold disk. |
| Reference Electrode | Provides a stable, known potential for the working electrode. | Ag/AgCl, Saturated Calomel (SCE). |
| Counter Electrode | Completes the circuit; typically made from inert wire. | Platinum wire. |
| Supporting Electrolyte | Carries current and minimizes resistive loss (iRu drop); must be electroinactive in the potential window of interest. | Tetraalkylammonium salts (e.g., TBAPF₆) in organic solvents; KCl in aqueous solutions. |
| Analyte | The redox-active species to be studied. | Ferrocene, pharmaceutical compounds, conductive polymers. |
| Solvent | Dissolves the electrolyte and analyte. | Acetonitrile (organic), Water (aqueous). |
Step-by-Step Procedure
- Solution Preparation: Prepare a solution containing the analyte (typically at concentrations of 1-10 mM) in a suitable solvent with a supporting electrolyte (typically 0.1 M) [9]. The supporting electrolyte concentration should be significantly higher than that of the analyte to ensure sufficient conductivity.
- Electrode Preparation: Clean the working electrode thoroughly according to the manufacturer's or established protocols (e.g., polishing on a microcloth with alumina slurry). Rinse the electrode with solvent and dry it [9].
- Setup: Place the solution into the electrochemical cell. Carefully insert the three electrodes into the cell, ensuring they are immersed in the solution but not touching each other.
- Instrument Configuration: On the potentiostat software, create a new cyclic voltammetry method. Set the parameters, which typically include:
- Initial Potential: The starting voltage (e.g., -0.4 V).
- High Vertex Potential: The most positive potential to be scanned.
- Low Vertex Potential: The most negative potential to be scanned.
- Scan Rate (ν): The rate at which the potential is changed (e.g., 0.1 V/s).
- Number of Scans: Often 2-3 scans are run to ensure a stable response.
- Data Acquisition: Initiate the scan. The potentiostat will control the potential and record the current, generating the cyclic voltammogram in real-time.
- Data Analysis: Once the scan is complete, use the software's analysis tools to mark the peak currents and peak potentials. Calculate the peak potential separation (ΔEp) and the peak current ratio (ipa/ipc) to assess reversibility [11]. Use the Randles-Sevcik equation for further quantitative analysis.
Advanced Application: Tafel Analysis for Kinetics
For electrochemically irreversible systems, Tafel analysis can be used to extract kinetic parameters such as the anodic (βa) and cathodic (βc) Tafel slopes, which are related to the electron transfer kinetics [12]. These are particularly important in fields like corrosion science and electrocatalysis.
Protocol: Generating a Tafel Plot from LSV Data
The following workflow outlines the process for transforming a Linear Sweep Voltammetry (LSV) segment into a Tafel plot using software like AfterMath.
Figure 2: Workflow for generating a Tafel plot and extracting Tafel slopes from LSV data [12].
- Data Preparation: Begin with a single-segment LSV. If your data is from a multi-segment CV, you must first extract the segment of interest (e.g., the forward scan) into a new LSV plot to preserve the original data [12].
- Axis and Unit Adjustment (Optional): The corrosion community often plots potential on the Y-axis and current density on the X-axis. Use a "Basic Math Operations" transform to swap the X and Y axes if needed. Convert current to current density by dividing the current values by the geometric area of the working electrode [12].
- Tafel Transformation: Apply a "Basic Math Operations" transform to the axis displaying current. Select the "Tafel log (|x|)" function, which calculates the base-10 logarithm of the absolute value of the current (or current density) [12]. The result is a Tafel plot (Potential vs. log |i|).
- Slope Determination: Use the software's "Baseline" tool to select the linear regions of the Tafel plot. The slope of the anodic branch is βa, and the slope of the cathodic branch is βc. These empirical values can be used in subsequent calculations, such as for corrosion rates in Linear Polarization Resistance (LPR) experiments [12].
The cyclic voltammogram serves as a fundamental fingerprint for redox-active species, providing deep insight into electrochemical behavior. By understanding its key features—the peak potentials, peak currents, and their relationships—researchers can determine the reversibility of a reaction, quantify kinetic parameters, and diagnose coupled chemical reactions. Adherence to standardized protocols for both basic CV experimentation and advanced data transformation, such as Tafel analysis, ensures the generation of robust, reproducible, and meaningful data. This is indispensable for advancing research in drug development, energy storage, materials science, and beyond.
The Nernst equation is a fundamental principle in electrochemistry that precisely relates the reduction potential of an electrochemical reaction to the standard electrode potential, temperature, and the activities (or concentrations) of the chemical species involved [13] [14]. For researchers utilizing cyclic voltammetry (CV), this equation provides the critical thermodynamic link between the measured potential in a voltammogram and the actual concentration of redox species at the electrode surface, enabling the quantification of reaction spontaneity and the determination of equilibrium constants [13] [15].
In the context of analyzing redox reactions, the Nernst equation describes the potential at which a redox couple exists at equilibrium at the electrode-solution interface. During a cyclic voltammetry experiment, where the electrode potential is linearly swept, the Nernst equation dictates how the relative concentrations of the oxidized (O) and reduced (R) forms of an analyte adjust instantaneously at the electrode surface to maintain equilibrium with the applied potential, assuming a reversible (fast) electron transfer process [16] [17]. This relationship is the cornerstone for interpreting the peak potentials and shapes of cyclic voltammograms, which in turn reveal vital information about the thermodynamics and kinetics of the system under study [18] [19].
Core Principles and Quantitative Relationships
The Fundamental Equation
The Nernst equation is derived from the relationship between the Gibbs free energy change under non-standard conditions and the electrical work that a cell can perform [13] [20]. For a general reversible redox reaction:
[ \text{O} + n\text{e}^- \rightleftharpoons \text{R} ]
The Nernst equation is expressed in its most general form as:
[ E = E^\circ - \frac{RT}{nF} \ln Q ]
or, for the specific reaction above:
[ E = E^\circ - \frac{RT}{nF} \ln \frac{a{\text{R}}}{a{\text{O}}} ]
where:
- ( E ) is the actual cell potential or half-cell potential (Volts, V)
- ( E^\circ ) is the standard cell potential or formal potential (V)
- ( R ) is the universal gas constant (8.314 J·mol⁻¹·K⁻¹)
- ( T ) is the absolute temperature (Kelvin, K)
- ( n ) is the number of electrons transferred in the redox reaction
- ( F ) is the Faraday constant (96,485 C·mol⁻¹)
- ( Q ) is the reaction quotient
- ( a{\text{R}} ) and ( a{\text{O}} ) are the chemical activities of the reduced and oxidized species, respectively [13] [14]
At room temperature (298.15 K or 25 °C), and converting from natural logarithm to base-10 logarithm, the equation simplifies to the widely used form:
[ E = E^\circ - \frac{0.0592}{n} \log \frac{[\text{R}]}{[\text{O}]} ]
Here, the activities are often approximated by concentrations (in mol·L⁻¹) for dilute solutions, a common condition in analytical experiments [13] [15]. This simplified version is exceptionally valuable for rapid, manual calculations.
Linking Potential, Free Energy, and Equilibrium
The Nernst equation is intrinsically connected to other key thermodynamic parameters, as summarized in the table below. These relationships allow researchers to extract comprehensive thermodynamic information from electrochemical measurements like cyclic voltammetry [13] [15].
Table 1: Thermodynamic Relationships in Electrochemistry
| Parameter | Mathematical Relationship | Significance in Redox Analysis |
|---|---|---|
| Standard Free Energy Change (( \Delta G^\circ )) | ( \Delta G^\circ = -nFE^\circ ) [15] | A negative ( \Delta G^\circ ) (positive ( E^\circ )) indicates a spontaneous reaction under standard conditions [15]. |
| Free Energy Change (( \Delta G )) | ( \Delta G = -nFE ) [13] [20] | The actual Gibbs energy change under non-standard conditions determines reaction spontaneity. |
| Equilibrium Constant (( K )) | ( \log K = \frac{nE^\circ}{0.0592} ) (at 298 K) [13] | Relates the standard cell potential directly to the thermodynamic equilibrium constant. A large ( K ) indicates the reaction proceeds far towards products [13] [15]. |
Table 2: Predicting Reaction Spontaneity from Potential and Composition
| Condition | Relationship | Reaction Spontaneity & Direction |
|---|---|---|
| Under Standard Conditions | ( Q = 1 ), ( E = E^\circ ) | If ( E^\circ > 0 ), reaction is spontaneous. If ( E^\circ < 0 ), reaction is non-spontaneous [15]. |
| At Equilibrium | ( Q = K ), ( E = 0 ) | No net reaction; the system is at equilibrium [13]. |
| Non-Standard Conditions | ( E = E^\circ - \frac{0.0592}{n} \log Q ) | If ( E > 0 ), reaction is spontaneous as written. If ( E < 0 ), reaction is spontaneous in the reverse direction [15]. |
Experimental Protocol: Applying the Nernst Equation in Cyclic Voltammetry
Research Reagent Solutions and Essential Materials
Table 3: Key Reagents and Materials for Cyclic Voltammetry
| Item | Function / Explanation |
|---|---|
| Potentiostat | Instrument that controls the potential between the working and reference electrodes and measures the resulting current [18]. |
| Three-Electrode System | Standard setup: a Working Electrode (e.g., glassy carbon, platinum) where the reaction of interest occurs, a Reference Electrode (e.g., Ag/AgCl) that provides a stable, known potential, and a Counter (Auxiliary) Electrode (e.g., platinum wire) that completes the circuit [9] [18]. |
| Supporting Electrolyte | A high-concentration, electrochemically inert salt (e.g., TBAPF₆, NaClO₄). Its primary function is to conduct current and minimize the effects of migratory mass transport, ensuring diffusion is the dominant mode of analyte transport [9]. |
| Redox-Active Analyte | The molecule or species under investigation (e.g., ferrocene, a quinone, a metal complex). It must be purified and of known, high purity for accurate quantitative analysis. |
| Solvent | A solvent suitable for electrochemical studies (e.g., acetonitrile, DMF, aqueous buffer). It must dissolve the analyte and electrolyte and have an appropriately wide electrochemical "window" where it is neither oxidized nor reduced within the potential range of interest [9]. |
Step-by-Step Methodology
Part A: Sample and Electrode Preparation
- Solution Preparation: Prepare a solution containing your redox-active analyte (typically 1-5 mM) in a suitable solvent. Add the supporting electrolyte at a significantly higher concentration (typically 0.1-0.2 M) to ensure sufficient conductivity [9].
- Electrode Preparation: Carefully polish the working electrode (e.g., a 3 mm diameter glassy carbon electrode) with successively finer alumina slurry (e.g., 1.0, 0.3, and 0.05 µm) on a microcloth pad. Rinse thoroughly with the purified solvent and then with the electrolyte solution to remove any polishing residue [18].
- Cell Assembly: Place the solution into a clean electrochemical cell. Insert the three electrodes, ensuring they are fully immersed. If necessary, purge the solution with an inert gas (e.g., nitrogen or argon) for at least 10-15 minutes to remove dissolved oxygen, which can interfere with the measurement, and maintain a gentle gas blanket over the solution during the experiment.
Part B: Instrument Setup and Data Acquisition
- Potentiostat Connection: Connect the working, reference, and counter electrodes to the corresponding leads of the potentiostat.
- Parameter Configuration: Open the control software and set up the cyclic voltammetry experiment with the following parameters [17] [19]:
- Initial Potential (Eᵢ): A potential where no faradaic current flows (e.g., 0 V vs. Ag/AgCl for ferrocene).
- Scan Range (Eλ): Set upper and lower switching potentials that comfortably bracket the expected redox event(s).
- Scan Rate (v): Begin with a moderate scan rate, such as 100 mV/s.
- Data Collection: Initiate the potential sweep. The potentiostat will apply the linear potential waveform and record the current response, generating a cyclic voltammogram (CV). For a reversible system, this will typically produce the characteristic "duck-shaped" CV with symmetrical oxidation and reduction peaks [9] [19].
Part C: Data Analysis Using the Nernst Equation
- Determine the Formal Potential (E°'): For a reversible couple, the formal potential (E°') is calculated as the midpoint between the anodic (Epa) and cathodic (Epc) peak potentials [19]: [ E^{\circ'} = \frac{E{pa} + E{pc}}{2} ]
- Verify Nernstian Behavior: Confirm the system is electrochemically reversible by checking that the peak separation (ΔEp = Epa - Epc) is close to 59/n mV at 25 °C and that the peak current ratio (Ipa/Ipc) is approximately 1 [17] [19].
- Calculate Concentration Ratios: Use the Nernst equation to determine the ratio of oxidized to reduced species at the electrode surface at any point during the potential sweep. For example, at the foot of the wave, you can calculate the tiny fraction of analyte that has been oxidized or reduced [16].
- Determine the Equilibrium Constant (K): If studying a coupled chemical equilibrium, use the formal potential shift to calculate the equilibrium constant for the chemical step using the relationships in Table 1 [13] [15].
Visualization of Core Concepts and Workflow
The Nernst Equation in a Cyclic Voltammetry Experiment
The following diagram illustrates how the Nernst equation governs the changing concentrations of redox species at the electrode surface during a cyclic voltammetry sweep, leading to the characteristic current response.
Experimental Workflow for CV-based Thermodynamic Analysis
This workflow outlines the key experimental and analytical steps for using cyclic voltammetry and the Nernst equation to determine thermodynamic parameters.
The Randles-Ševčík equation is a fundamental principle in electrochemistry that quantitatively describes the peak current response in cyclic voltammetry (CV) experiments for reversible, diffusion-controlled redox reactions [21] [22]. This equation provides a critical link between experimentally measurable parameters (peak current) and intrinsic properties of the electroactive species, such as its diffusion coefficient [23]. For researchers in redox reaction analysis and drug development, it serves as an indispensable tool for quantifying electrochemical processes, characterizing new compounds, and verifying experimental conditions [24].
The equation was independently derived in 1948 by John Edward Brough Randles and Antonín Ševčík during post-war advancements in electroanalytical techniques, which shifted focus from steady-state polarography to dynamic studies of redox kinetics [22]. Its development enabled quantitative analysis of electrochemical systems without complex numerical simulations, making it a cornerstone technique that remains widely applied in materials science, biosensor development, and pharmaceutical research [22].
Theoretical Foundation
Mathematical Formulations
The Randles-Ševčík equation is derived from Fick's laws of diffusion under conditions where electron transfer kinetics are rapid relative to mass transport (electrochemically reversible systems) [21] [22]. The general form of the equation is:
$$i_p = 0.4463 \, nFAC \left( \frac{nF \nu D}{RT} \right)^{1/2}$$
For practical applications at standard laboratory temperature (25°C), the equation simplifies to:
$$i_p = (2.69 \times 10^5) \, n^{3/2} A D^{1/2} C \nu^{1/2}$$
The following table details all parameters and their units required for applying the Randles-Ševčík equation.
Table 1: Parameters in the Randles-Ševčík Equation
| Parameter | Symbol | Units | Description |
|---|---|---|---|
| Peak Current | (i_p) | Amperes (A) | Maximum current observed during potential sweep |
| Number of Electrons | (n) | Dimensionless | Electrons transferred in redox event |
| Electrode Area | (A) | cm² | Electroactive surface area of working electrode |
| Diffusion Coefficient | (D) | cm²/s | Measure of species mobility in solution |
| Concentration | (C) | mol/cm³ | Bulk concentration of electroactive species |
| Scan Rate | (\nu) | V/s | Rate of potential sweep |
| Faraday Constant | (F) | C/mol | Electrical charge per mole of electrons (96485 C/mol) |
| Gas Constant | (R) | J/(mol·K) | Universal gas constant (8.314 J/(mol·K)) |
| Temperature | (T) | K | Absolute temperature |
Diagnostic Applications for Reaction Mechanisms
The relationship (i_p \propto \nu^{1/2}) provides critical diagnostic power for distinguishing reaction mechanisms. A linear plot of peak current versus the square root of scan rate indicates a diffusion-controlled process with freely diffusing species [23] [25]. Deviations from this linearity suggest alternative mechanisms:
- Surface-adsorbed species: Electron transfer occurs through molecules attached to the electrode surface rather than freely diffusing [25].
- Quasi-reversible or irreversible kinetics: Electron transfer rates become slow relative to the scan rate [24].
- Electrochemical irreversibility: The redox reaction is not reversible, invalidating the equation's assumptions [25].
For quasi-reversible systems (typically 63 mV < nΔEp < 200 mV), a modified Randles-Ševčík equation incorporating a dimensionless kinetic parameter K(Λ,α) must be used [24]:
$$I_p = (2.69 × 10^5 \, n^{3/2} A D C \nu^{1/2}) K(Λ,α)$$
The following diagram illustrates the diagnostic workflow for interpreting cyclic voltammetry data using the Randles-Ševčík equation.
Experimental Protocols
Determining Diffusion Coefficients
Purpose: Calculate the diffusion coefficient (D) of an electroactive species using the Randles-Ševčík equation [21] [23].
Materials:
- Standard redox probe with known behavior (e.g., 1.0 mM potassium ferricyanide in 1.0 M KCl)
- Supporting electrolyte appropriate for your system
- Three-electrode electrochemical cell
- Potentiostat with cyclic voltammetry capability
- Working electrode with known electroactive area
Procedure:
- Prepare an electrochemical cell containing your analyte at known concentration in supporting electrolyte [24].
- Record cyclic voltammograms at multiple scan rates (e.g., 10, 25, 50, 100, 200, 400 mV/s) [24].
- For each voltammogram, measure the peak current (ip) for the oxidation or reduction wave.
- Plot ip versus ν1/2.
- Perform linear regression on the data. The slope (m) of this plot relates to the diffusion coefficient:
$$D = \left( \frac{\text{slope}}{2.69 \times 10^5 \, n^{3/2} A C} \right)^2$$
Validation: The plot of ip versus ν1/2 should be linear with a correlation coefficient (R²) >0.995, and the peak potential separation (ΔEp) should be close to 59/n mV for a reversible system [24].
Calculating Electroactive Surface Area
Purpose: Determine the electroactive area (A) of a working electrode, which often differs from its geometric area [23] [24].
Materials:
- Standard redox couple with known diffusion coefficient (e.g., 1.0 mM ferrocene in acetonitrile with 0.1 M TBAPF6 as supporting electrolyte)
- Supporting electrolyte
- Electrochemical cell and potentiostat
- Electrode to be characterized
Procedure:
- Record cyclic voltammograms of the standard solution at multiple scan rates.
- Measure the peak current (ip) for each scan rate.
- Plot ip versus ν1/2 and determine the slope.
- Calculate the electroactive area using:
$$A = \frac{\text{slope}}{2.69 \times 10^5 \, n^{3/2} D^{1/2} C}$$
Applications: This protocol is essential for characterizing modified electrodes, assessing electrode fouling, and validating electrode cleaning procedures [23] [24].
Research Reagent Solutions
The following table outlines essential materials and their functions for experiments utilizing the Randles-Ševčík equation.
Table 2: Essential Research Reagents and Materials
| Material/Reagent | Function/Application | Example Specifications |
|---|---|---|
| Standard Redox Probes | Reference compounds for method validation | 1-5 mM potassium ferricyanide, ferrocene, or ruthenium hexamine [24] |
| Supporting Electrolyte | Minimize migration effects, provide conductivity | 0.1-1.0 M KCl, TBAPF6, or other salts [24] |
| Working Electrodes | Platform for redox reactions | Glassy carbon, gold, or platinum electrodes [26] [23] |
| Potentiostat | Instrument for applying potential and measuring current | Capable of cyclic voltammetry with adjustable scan rates [27] |
| Solvents | Dissolve analytes and electrolytes | Acetonitrile, water, DMF; purified and deoxygenated [24] |
Applications in Pharmaceutical and Analytical Research
Antioxidant Capacity Assessment
Cyclic voltammetry combined with the Randles-Ševčík equation provides a powerful method for evaluating antioxidant potential in natural products and pharmaceuticals [28] [29]. Recent studies have successfully correlated anodic current measurements with traditional antioxidant assays (DPPH, ABTS), offering insights into electron-donating capabilities of bioactive compounds [28]. The peak current directly relates to antioxidant concentration and strength, while the peak potential indicates the reducing power [28]. This approach has been applied to characterize vegetable extracts, protein hydrolysates, and medicinal plants, supporting drug development from natural sources [29].
Characterization of Complex Systems
The equation facilitates quantitative analysis of complex interactions relevant to pharmaceutical sciences. Research on mercuric chloride interactions with Orange G dye demonstrated how scan rate studies combined with the Randles-Ševčík relationship can elucidate complexation behavior and determine stability constants [26]. Such approaches help understand how toxic compounds interact with biological molecules, contributing to environmental monitoring and pharmaceutical safety assessments [26].
Advanced Applications and Recent Developments
Recent methodological advances continue to expand the equation's applications. Novel techniques like opto-iontronic microscopy now enable monitoring electrochemical processes at nanoscale volumes, validating theoretical models including the Randles-Ševčík relationship in confined environments [30]. Such developments open possibilities for high-sensitivity analysis with potential applications in single-molecule electrochemistry relevant to drug discovery [30].
Critical Considerations and Method Validation
System Requirements and Limitations
Researchers must verify key assumptions before applying the Randles-Ševčík equation:
- Reversibility: The redox system must be electrochemically reversible (fast electron transfer kinetics) [21] [22].
- Diffusion control: Mass transport must occur primarily through diffusion, not convection or adsorption [25].
- No competing reactions: The system should contain only the redox reaction of interest [21].
- Planar electrode: The equation assumes semi-infinite linear diffusion to a planar electrode surface [22].
Troubleshooting Common Issues
The following workflow diagram outlines a systematic approach for diagnosing and addressing common problems in Randles-Ševčík analysis.
Data Interpretation Guidelines
For reliable results, researchers should:
- Use at least five different scan rates covering at least an order of magnitude (e.g., 10-500 mV/s) [24].
- Ensure consistent temperature control, as diffusion coefficients are temperature-dependent.
- Verify electrode cleanliness between experiments, as fouling significantly affects electroactive area [23].
- Confirm the absence of background currents that could interfere with peak current measurements.
- Validate findings with complementary techniques when characterizing new systems [24].
When properly applied and validated, the Randles-Ševčík equation provides robust quantitative analysis of redox systems, contributing significantly to pharmaceutical development, materials characterization, and fundamental electrochemical research.
Distinguishing Reversible, Irreversible, and Quasi-reversible Electron Transfer Processes
In the analysis of redox reactions using cyclic voltammetry (CV), categorizing the nature of electron transfer is a fundamental step in interpreting electrochemical data and understanding underlying reaction mechanisms. The terms reversible, irreversible, and quasi-reversible describe the kinetic facility of electron transfer between the electrode and electroactive species [31] [32]. For researchers in drug development, accurately distinguishing these processes is critical, as electron transfer kinetics can influence the stability, reactivity, and redox properties of pharmaceutical compounds. A reversible process indicates fast electron transfer kinetics where the redox couple rapidly establishes equilibrium at the electrode surface at each potential. In contrast, an irreversible process features slow electron transfer, requiring significant overpotential to drive the reaction. The quasi-reversible category encompasses an intermediate regime where both electron transfer kinetics and mass transport influence the voltammetric response [31]. This application note provides a structured framework for distinguishing these electron transfer processes through defined diagnostic parameters and experimental protocols.
Theoretical Foundations and Diagnostic Criteria
Fundamental Definitions
Electrochemical Reversibility: This concept specifically refers to the kinetics of heterogeneous electron transfer at the electrode-solution interface [32]. A system is considered electrochemically reversible when the electron transfer rate is sufficiently high to maintain Nernstian equilibrium at the electrode surface throughout the potential scan [31]. This is distinct from chemical reversibility, which concerns the stability of the redox-generated species to subsequent chemical reactions [32].
Chemical Reversibility: A system is chemically reversible if the electrogenerated species (e.g., the reduced form "Red" produced from the oxidized form "Ox") is stable on the experimental timescale and can be converted back to its original form during the reverse potential scan [32]. When the product undergoes a subsequent irreversible chemical reaction (e.g., R → Z), the system is deemed chemically irreversible, often manifesting as the disappearance of the return peak in CV [31] [33].
Quantitative Diagnostic Parameters
The following parameters, derived from analysis of cyclic voltammograms, serve as primary diagnostics for classifying electron transfer processes.
Table 1: Diagnostic Criteria for Classifying Electron Transfer Processes in Cyclic Voltammetry
| Parameter | Reversible | Quasi-Reversible | Irreversible |
|---|---|---|---|
| Peak Separation (ΔEp) | ≈ 59/n mV (at 25°C) [33] | > 59/n mV [33] | > 59/n mV, significantly larger [31] |
| Scan Rate Dependence of Ep | Constant [34] | Shifts with scan rate [31] | Shifts with scan rate; linear with log(ν) [34] |
| Peak Current Ratio (ipa/ipc) | ≈ 1 [33] | Near 1 (but shape changes) [31] | Deviates from 1 [31] |
| Current Function (ip/ν1/2) | Constant [33] | Decreases with increasing ν [31] | Varies, lower magnitude |
| Rate Constant, k° (cm/s) | Large (> ~0.1-1 cm/s) [33] | Intermediate (~10-5 to 10-1) [31] | Small (< ~10-5) [31] |
Conceptual Workflow for Classification
The following diagram illustrates the logical decision process for classifying an electron transfer process based on cyclic voltammetry data.
Figure 1: Decision workflow for classifying electron transfer processes from CV data.
Experimental Protocols for Distinction
Protocol 1: Multi-Scan Rate CV Analysis
Purpose: To determine the effect of scan rate on peak potential and current, which is crucial for classifying electron transfer reversibility [34].
Materials:
- Potentiostat with standard three-electrode cell setup
- Working electrode (e.g., glassy carbon, Pt disk)
- Reference electrode (e.g., Ag/AgCl, SCE)
- Counter electrode (Pt wire)
- Electrolyte solution (e.g., 0.1 M KNO3, PBS)
- Analyte of interest (e.g., 1-5 mM concentration)
Procedure:
- Prepare the electrochemical cell with analyte dissolved in supporting electrolyte.
- Polish the working electrode (if solid) to a mirror finish using alumina slurry (1.0, 0.3, and 0.05 µm sequentially) and rinse thoroughly with deionized water [34].
- Set the initial potential at least 200 mV more positive (for reduction) or negative (for oxidation) than the expected formal potential (E°).
- Define the switching potential at least 200 mV beyond the observed peak potential.
- Record CVs at a series of scan rates (e.g., 10, 25, 50, 100, 250, 500 mV/s).
- For each voltammogram, measure the anodic peak potential (Epa), cathodic peak potential (Epc), anodic peak current (ipa), and cathodic peak current (ipc).
Data Analysis:
- Calculate ΔEp = Epa - Epc for each scan rate.
- Plot Ep versus log(ν) for both anodic and cathodic peaks.
- Plot ip versus ν1/2 to verify diffusion control.
- Plot log(ip) versus log(ν); the slope should be approximately 0.5 for diffusion-controlled processes.
Interpretation:
- If ΔEp is close to 59/n mV and independent of scan rate, the system is reversible [34].
- If ΔEp > 59/n mV and increases with scan rate, and Ep shifts linearly with log(ν) with a slope of approximately ±60/αn mV/decade, the system is irreversible [34].
- For quasi-reversible systems, ΔEp is greater than 59/n mV and increases with scan rate, but the peaks remain well-defined [31].
Protocol 2: Determining Heterogeneous Electron Transfer Rate Constant (k°)
Purpose: To quantify the standard heterogeneous electron transfer rate constant, which provides a numerical basis for classifying electron transfer processes [35].
Materials: Same as Protocol 1, with emphasis on careful control of experimental conditions.
Procedure:
- Follow steps 1-4 from Protocol 1, ensuring excellent electrode preparation and ohmic drop compensation.
- For reversible systems, k° can be estimated from the scan rate (νrev) at which the system begins to deviate from reversibility using the relationship: k° ≈ 0.3(νrevnF/RT)1/2 [33].
- For quasi-reversible systems, record CVs across a wide range of scan rates (e.g., 0.01 to 100 V/s if accessible).
- Use square-wave voltammetry (SWV) as a complementary technique by scanning frequency and amplitude, then fitting data to a model incorporating Butler-Volmer kinetics [35].
Data Analysis using Numerical Simulation:
- Model the electrochemical cell using Fick's second law of diffusion: ∂CO/∂t = DO∇2CO and ∂CR/∂t = DR∇2CR [35].
- Apply Butler-Volmer kinetics as a boundary condition at the electrode surface: Flux = -k°[e-αnF/RT(E-E°)CO(0,t) - e(1-α)nF/RT(E-E°)CR(0,t)] [35].
- Iteratively adjust k° and α in simulations to minimize the difference between simulated and experimental voltammograms.
Classification Criteria:
- Reversible: k° > 0.1-1 cm/s [33]
- Quasi-reversible: 10-5 < k° < 0.1 cm/s [31]
- Irreversible: k° < 10-5 cm/s [31]
Experimental Workflow for Comprehensive Analysis
The complete experimental pathway for characterizing electron transfer processes is illustrated below.
Figure 2: Comprehensive experimental workflow for characterizing electron transfer processes.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 2: Key Research Reagent Solutions and Materials for Electron Transfer Studies
| Item | Function/Application | Example Specifications |
|---|---|---|
| Supporting Electrolyte | Provides ionic conductivity; minimizes ohmic drop; controls ionic strength | 0.1 M KNO3, PBS buffer, TBAPF6 in organic solvents |
| Standard Redox Couples | System validation and calibration | 1.0 mM K3Fe(CN)6 in 1.0 M KNO3 (reversible benchmark) [33] |
| Electrode Polishing Kit | Ensines reproducible electrode surface morphology | Alumina slurry (1.0, 0.3, 0.05 µm); polishing pads |
| Potentiostat | Applies potential waveform and measures current | Commercial instrument with scan rates from 0.1 mV/s to 10,000 V/s |
| Faradaic Cage | Minimizes external electromagnetic interference | Enclosed metal enclosure grounded to potentiostat |
| Solvent Systems | Dissolves analytes of varying polarity | Acetonitrile (non-aqueous), Water (aqueous), DMF |
| Numerical Simulation Software | Extracts kinetic parameters from voltammetric data | DIGISIM, COMSOL, or custom finite-difference algorithms [35] |
Advanced Applications and Considerations
Square-Wave Voltammetry for Kinetic Analysis
For particularly challenging systems with very fast or very slow electron transfer, square-wave voltammetry (SWV) provides enhanced sensitivity for kinetic analysis [35]. This technique applies a series of square-wave pulses superimposed on a staircase ramp, effectively discriminating against capacitive currents. The relationship between square-wave frequency and peak current provides quantitative information about electron transfer rates, extending the measurable range of k° values beyond what is accessible through CV alone [35]. The numerical simulation approach described in Protocol 2 can be adapted for SWV data by modeling the more complex potential waveform.
Distinguishing Chemical vs. Electrochemical Irreversibility
A critical challenge in interpreting irreversible voltammetric responses is distinguishing between slow electron transfer kinetics (electrochemical irreversibility) and rapid chemical reaction of the electrogenerated species (chemical irreversibility) [31] [32]. This distinction has significant implications in drug development, where chemical irreversibility may indicate metabolic instability or reactive metabolite formation.
Diagnostic Approach:
- Systematic scan rate studies: In chemically irreversible systems following an EC mechanism (Electron transfer followed by Chemical step), the ratio of reverse-to-forward peak currents (ip,rev/ip,fwd) decreases with decreasing scan rate, as the chemical step has more time to consume the electrogenerated species [33].
- Controlled potential electrolysis with product analysis: Bulk electrolysis at the peak potential followed by analytical characterization (e.g., HPLC, NMR) of the solution can identify decomposition products.
- Variable time scale experiments: Using ultramicroelectrodes to access shorter experimental time scales can sometimes reveal reversibility that is masked at conventional time scales.
Accurate classification of electron transfer processes as reversible, quasi-reversible, or irreversible provides fundamental insights into redox behavior that is essential for research in electrochemistry, materials science, and drug development. The protocols and diagnostic criteria outlined in this application note establish a systematic approach for distinguishing these processes through multi-scan rate cyclic voltammetry, complemented by numerical simulation to extract quantitative kinetic parameters. For researchers in pharmaceutical development, this classification not only characterizes electron transfer kinetics but also reveals potential chemical reactivity of redox-generated species, informing drug stability and metabolic fate predictions. The experimental workflows and decision trees presented here offer a standardized methodology applicable across diverse research domains where understanding electron transfer is critical.
The Electric Double Layer (EDL) and Its Role in Electrode-Solution Interfaces
The Electrical Double Layer (EDL) is a fundamental concept in electrochemistry, describing the structured arrangement of ions and molecules that forms at the interface between an electrode and an electrolyte solution. This region is critical because its properties govern the reactivity, capacitance, and electron-transfer kinetics of electrochemical processes. When a charged electrode is immersed in an electrolyte, ions from the solution arrange themselves to screen the surface charge. This creates a complex interface consisting of a compact layer of strongly adsorbed ions (the Stern layer) and a diffuse layer where ions are mobile, influenced by both electrostatic forces and diffusion [36]. A detailed understanding of the EDL is indispensable for interpreting electrochemical techniques, notably cyclic voltammetry (CV), a cornerstone method for analyzing redox reactions. The structure and dynamics of the EDL directly influence key CV parameters, such as peak currents, peak potentials, and the overall shape of the voltammogram, thereby providing crucial insights into reaction thermodynamics and kinetics [37] [36].
Theoretical Foundations of the EDL
The classical Gouy-Chapman-Stern (GCS) model provides a foundational, though simplified, description of the EDL. This model partitions the interface into two main regions: the inner Stern layer (or Helmholtz layer), comprising ions specifically adsorbed and immobilized on the electrode surface, and the outer diffuse layer, where a cloud of mobile ions screens the remaining surface charge [36]. The entire EDL can be electrically represented as a capacitance, often modeled as the Stern capacitance and the diffuse layer capacitance acting in series [36].
However, advanced computational studies reveal that the real picture is more complex. The EDL is not a simple mean-field structure but is highly dependent on molecular-scale interactions. For instance, at metal oxide-electrolyte interfaces, the surface charge is not uniform but is determined by the protonation and deprotonation of specific surface sites, which is highly sensitive to the pH of the solution relative to the point of zero charge (pHPZC) [38]. Ab initio machine learning potential simulations have shown that the charging mechanisms can differ significantly under acidic versus basic conditions, leading to distinct capacitive behaviors [38]. Furthermore, the properties of the first few layers of water at the interface deviate substantially from bulk water, a nuance that continuum models like GCS cannot capture [38]. These molecular-scale insights are crucial for a accurate interpretation of electrochemical data.
Quantitative EDL Data from Experimental and Simulation Studies
The following tables summarize key quantitative findings on EDL properties from recent research, highlighting the impact of material, solution conditions, and measurement technique.
Table 1: EDL Capacitance from Various Studies
| Electrode Material | Electrolyte | pH (vs. pHPZC) | Capacitance | Measurement Technique | Source |
|---|---|---|---|---|---|
| Anatase TiO₂ | 0.4 M NaCl | Acidic (pH < pHPZC) | ~7.69 µC/cm² (Surface Charge) | DPLR Molecular Simulation | [38] |
| Anatase TiO₂ | 0.4 M NaCl | Basic (pH > pHPZC) | ~7.54 µC/cm² (Surface Charge) | DPLR Molecular Simulation | [38] |
| Planar Electrode | Aqueous Solution | N/A | Constant at low scan rates | CV with MPNP Model | [36] |
Table 2: Impact of EDL on Redox Kinetics (Fe(CN)₆³⁻/⁴⁻ System)
| Electrode Structure | EDL Characteristics | Electron Transfer Kinetics | Observed Peak Potential Separation (ΔEₚ) | Source |
|---|---|---|---|---|
| Ag Monolayer on Au | Similar to bulk Ag EDL | Corresponds to Au electrode | Standard for a reversible system | [37] |
| Ag Multilayer on Au | Forms Ag Hexacyanoferrate (II) film | Altered by ohmic film resistance | Increases with number of Ag layers | [37] |
Experimental Protocols
This section provides detailed methodologies for investigating the EDL using cyclic voltammetry and an advanced optical technique.
Protocol: Probing EDL Effects Using a Model Redox Couple
This protocol uses the Ferricyanide/Ferrocyanide (Fe(CN)₆³⁻/⁴⁻) redox couple to characterize the EDL and electron transfer kinetics on modified electrodes [37].
Research Reagent Solutions
- Supporting Electrolyte: 1 M KNO₃ in triply-distilled water.
- Redox Probe: Potassium ferricyanide (K₃Fe(CN)₆) in the supporting electrolyte.
- Electrode Cleaning Solution: 0.5 M H₂SO₄.
- Electrode Modification Electrolyte: 0.1 M NaF, adjusted to pH 5 with sulfuric acid.
- Silver Plating Solution: Ag⁺ ions in the NaF electrolyte.
Procedure
- Working Electrode Preparation: Begin with a polycrystalline gold wire working electrode (e.g., 0.7 mm diameter). Polish the electrode mechanically with 0.3 µm alumina powder. Subsequently, electro-polish in 0.5 M H₂SO₄ to achieve a pristine surface [37].
- Electrode Modification (Silver Deposition):
- Immerse the clean Au working electrode in the Ag⁺-containing NaF electrolyte (pH 5).
- Use an underpotential deposition protocol to deposit a monolayer of silver atoms. For multilayer deposits, continue the electrodeposition process.
- Characterize the deposit using techniques like Scanning Electron Microscopy (SEM) to confirm surface coverage and morphology [37].
- Cyclic Voltammetry Measurement:
- Transfer the modified electrode to an electrochemical cell containing the Fe(CN)₆³⁻/⁴⁻ solution in 1 M KNO₃.
- Use a standard three-electrode setup with a Pt counter electrode and a suitable reference electrode (e.g., Ag/AgCl).
- Record cyclic voltammograms over a suitable potential window (e.g., -0.2 V to 0.5 V vs. Ag/AgCl) at multiple scan rates (e.g., from 10 mV/s to 500 mV/s) [37].
- Data Analysis:
- Kinetics Analysis: For a monolayer Ag deposit, the EDL structure is like bulk silver, but the electron transfer kinetics will still reflect the underlying gold substrate [37].
- Ohmic Effects Analysis: For multilayer Ag deposits, an increase in the peak potential separation (ΔEₚ) with deposit thickness is not due to slowed kinetics but to the increased ohmic resistance of a formed silver hexacyanoferrate (II) film. This can be confirmed with the Randles-Sevcik equation; a linear relationship between peak current and the square root of scan rate confirms a diffusion-controlled process despite the resistive layer [37].
Protocol: Opto-iontronic Microscopy for Nanoscale EDL Dynamics
This advanced protocol leverages optical microscopy to directly monitor EDL charging and coupled redox reactions within nanoconfined volumes, providing unprecedented spatial resolution [30].
Research Reagent Solutions
- Electrolyte and Redox Probe: 1,1-Ferrocenedimethanol (Fc(MeOH)₂) dissolved in 100 mM KCl aqueous solution.
- Nanohole Fabrication Material: A 100 nm thin gold film supported on a glass substrate.
Procedure
- Nanofabrication: Fabricate an array of nanoholes in the gold film using a Focused Ion Beam (FIB). Typical nanohole dimensions are 100 nm in depth and 75-100 nm in diameter, creating an attoliter-scale measurement volume [30].
- Optical Setup:
- Employ a Total Internal Reflection (TIR) illumination setup. Illuminate the glass-gold interface with a laser to generate an evanescent field that penetrates into the nanoholes without directly illuminating the bulk solution.
- Use a high-numerical-aperture objective to collect the scattered light from the nanoholes [30].
- Electro-Optical Measurement:
- Connect the gold film as the working electrode in a standard potentiostat setup.
- Apply a cyclic voltammetry potential sweep (e.g., from -0.2 V to 0.2 V) to the nanohole electrode while simultaneously recording the optical scattering signal.
- Use a lock-in amplifier synchronized to a small potential modulation to significantly enhance the signal-to-noise ratio and detect minute optical changes caused by the electrochemical processes [30].
- Data Analysis:
- The optical scattering intensity is linked to the local ion concentration within the nanohole. Compare the experimentally measured optical signal with theoretical predictions from a Poisson-Nernst-Planck-Butler-Volmer (PNP-BV) model.
- The correlation between the optical contrast and the modeled ion concentration provides a direct, label-free readout of the electrochemical reaction dynamics and EDL (dis)charging within the nanoconfined space [30].
Visualization of EDL Structure and Experimental Workflows
Diagram 1: This visualization illustrates the structure of the Electrical Double Layer (EDL) at a positively charged electrode. The Stern Layer contains specifically adsorbed ions and water molecules. The Diffuse Layer consists of a cloud of mobile cations and anions, the distribution of which is governed by a balance between electrostatic attraction and thermal motion. The structure and dynamics of this entire interface control electrochemical reactivity [38] [36].
Diagram 2: This workflow outlines the key steps in a protocol to investigate the EDL and its effects on redox reactions using cyclic voltammetry. The process involves meticulous electrode preparation, surface modification, electrochemical measurement, and data analysis to extract parameters that reveal the properties of the interface [37] [9].
Practical CV Methods and Applications in Drug Development and Research
Cyclic Voltammetry (CV) is a powerful and ubiquitous electrochemical technique used to study redox reaction mechanisms, providing both qualitative and quantitative information about electrochemical systems [39]. In pharmaceutical and diagnostic research, CV enables the investigation of electron transfer processes crucial for understanding drug metabolism, biomarker detection, and biosensor development [40]. This technique involves sweeping the working electrode potential linearly with time between specified limits while measuring the resulting current, generating a characteristic "duck-shaped" plot known as a voltammogram [9]. The resulting current-potential data reveals crucial electrochemical parameters including formal potentials, electron transfer kinetics, diffusion coefficients, and reaction mechanisms [11]. This application note provides a standardized protocol for researchers establishing CV methodologies for redox reaction analysis, with particular emphasis on proper electrolyte preparation, instrument configuration, and measurement execution to ensure reproducible and meaningful results.
Theoretical Principles and Key Parameters
Fundamental Electrochemical Theory
In CV, a three-electrode system subjects the electrochemical cell to a linearly cycled potential sweep while measuring the current response [9]. For a reversible redox couple, the peak current (ip) is described by the Randles-Ševčík equation at 25°C:
[i_p = (2.69 \times 10^5) \cdot n^{3/2} \cdot A \cdot D^{1/2} \cdot C \cdot v^{1/2}]
where n is the number of electrons transferred, A is the electrode area (cm²), D is the diffusion coefficient (cm²/s), C is the concentration (mol/cm³), and v is the scan rate (V/s) [9]. The peak potential separation (ΔEp = Epa - Epc) for a reversible, one-electron transfer process is approximately 59 mV at 25°C, with equal anodic and cathodic peak currents (ipa/ipc = 1) [11]. Reversibility requires fast electron transfer kinetics sufficient to maintain Nernstian equilibrium conditions throughout the potential scan [11].
Criteria for Electrochemical Reversibility
Electrochemical reversibility depends on the relative values of the standard heterogeneous electron transfer rate constant (ks) and the scan rate (v) [11]. A system exhibits reversible behavior when ks/v is sufficiently large to maintain Nernstian surface concentrations. Quasi-reversible systems show ΔEp > 59/n mV, with values increasing with scan rate, while irreversible systems display shifted peak potentials and diminished reverse peaks [11]. Chemical reactions coupled to electron transfer, such as acid-base reactions or decomposition processes, can also cause irreversibility by altering the redox species during the potential cycle [41].
Table 1: Diagnostic Parameters for Reversible Redox Systems in Cyclic Voltammetry
| Parameter | Reversible System Criteria | Experimental Significance |
|---|---|---|
| Peak Potential Separation (ΔEp) | 59.2/n mV at 25°C | Indicates thermodynamic reversibility and number of electrons transferred |
| Peak Current Ratio (ipa/ipc) | 1 at all scan rates | Confirms stability of redox species during potential cycle |
| Peak Current Function (ip/v¹/²) | Independent of scan rate | Validates diffusion-controlled process |
| Peak Potential | Independent of scan rate | Suggests fast electron transfer kinetics |
Materials and Reagent Preparation
Research Reagent Solutions and Materials
Table 2: Essential Reagents and Materials for Cyclic Voltammetry Experiments
| Item | Specification | Function/Purpose |
|---|---|---|
| Supporting Electrolyte | KCl, PBS (0.1-1.0 M) | Provides ionic conductivity, controls ionic strength |
| Redox Probe | Potassium ferricyanide, Ferrocene, [Ru(NH₃)₆]³⁺ | Generates faradaic current for redox process characterization |
| Solvent | Acetonitrile, Aqueous buffers | Dissolves electrolyte and redox species |
| Working Electrode | Glassy carbon, Gold, Platinum | Surface for redox reactions to occur |
| Reference Electrode | Ag/AgCl, SCE | Provides stable potential reference |
| Counter Electrode | Platinum wire, Graphite rod | Completes electrical circuit without reaction interference |
| Purification Gas | Nitrogen, Argon | Removes dissolved oxygen from solution |
Electrolyte and Redox Probe Selection
The choice of electrolyte composition significantly impacts redox reactivity and electron transfer kinetics [42] [40]. Phosphate Buffered Saline (PBS) provides pH stabilization but may yield lower sensitivity compared to potassium chloride (KCl) at equivalent ionic strengths [40]. For the ferro/ferricyanide system ([Fe(CN)₆]³⁻/⁴⁻), increased electrolyte ionic strength shifts the RC semicircle in Nyquist plots to higher frequencies, enhancing signal response [40]. However, [Fe(CN)₆]³⁻/⁴⁻ exhibits surface-sensitive behavior on carbon electrodes and may show quasi-reversible kinetics, while [Ru(NH₃)₆]³⁺/²⁺ behaves as a more ideal outer-sphere redox probe but at higher cost [43]. Optimal signal-to-noise ratios for low-cost analyzers can be achieved using buffered electrolytes like PBS with high ionic strength and lowered redox probe concentrations [40].
Experimental Setup and Protocols
Electrolyte Preparation Protocol
Solution Preparation: Prepare supporting electrolyte (e.g., 0.1 M KCl or PBS) using high-purity water (resistivity ≥18 MΩ·cm). Accurately weigh electrolyte salts using analytical balance and dissolve in appropriate solvent volume [43].
Redox Probe Addition: Add redox-active species to electrolyte solution. Typical concentrations range from 1-5 mM for routine characterization. For ferricyanide, prepare 5 mM K₃[Fe(CN)₆] in 0.1 M KCl [43].
Oxygen Removal: Sparge solution with inert gas (N₂ or Ar) for 10-15 minutes before measurements to remove dissolved oxygen, which can interfere with redox processes [44].
pH Adjustment: Adjust pH using dilute acid/base solutions as needed. For PBS, maintain pH 7.4 for biological applications [40].
Electrode Preparation and Cell Assembly
Working Electrode Polishing: Polish glassy carbon electrode sequentially with 1.0, 0.3, and 0.05 μm alumina slurry on microcloth pads. Rinse thoroughly with purified water between polishing steps [44].
Electrode Cleaning: Sonicate electrode in appropriate solvent (water, ethanol) for 2-5 minutes to remove residual polishing material [44].
Cell Assembly: Insert clean, dry electrodes into cell ports, ensuring proper orientation and immersion depth. Connect electrodes to potentiostat following manufacturer's configuration [45].
Solution Transfer: Transfer deoxygenated electrolyte solution to electrochemical cell, ensuring electrodes are fully immersed. Maintain inert atmosphere during measurement if needed [44].
Potentiostat Configuration and Measurement
Instrument Warm-up: Switch on potentiostat at least 30 minutes before measurements to ensure thermal stability and accurate readings [44].
Open Circuit Potential Measurement: Measure open circuit potential (Eoc) to establish baseline potential before applying controlled potentials [45].
Parameter Settings: Configure CV parameters based on experimental requirements:
- Initial potential: Typically 100-200 mV before expected E⁰'
- Vertex potentials: Set to encompass redox events of interest
- Scan rate: 10-100 mV/s for initial characterization
- Number of cycles: 2-5 cycles to ensure reproducibility [45]
Current Range Selection: Select appropriate current range based on expected response. Use autoranging if available, or estimate using Randles-Ševčík equation [45].
Experiment Execution: Initiate measurement sequence, monitoring real-time voltammogram display for anomalies. Save data in appropriate format for subsequent analysis [39].
Cyclic Voltammetry Experimental Workflow
Data Analysis and Interpretation
Voltammogram Analysis Protocol
Peak Identification: Identify anodic (Epa) and cathodic (Epc) peak potentials and corresponding currents (ipa, ipc) from the voltammogram [11].
Reversibility Assessment: Calculate ΔEp = Epa - Epc and ipa/ipc ratio. Compare to theoretical values for reversible systems (ΔEp = 59/n mV, ipa/ipc = 1) [11].
Formal Potential Determination: Calculate formal potential E⁰' = (Epa + Epc)/2 for reversible systems [11].
Scan Rate Studies: Perform CV at multiple scan rates (e.g., 10-1000 mV/s). Plot ip vs. v¹/² to verify linear relationship expected for diffusion-controlled processes [9].
Electroactive Area Calculation: Using the Randles-Ševčík equation with known concentration and diffusion coefficient, calculate electroactive area from slope of ip vs. v¹/² plot [9].
Table 3: Troubleshooting Common Cyclic Voltammetry Issues
| Problem | Potential Causes | Solutions |
|---|---|---|
| Large ΔEp (>59/n mV) | Slow electron transfer kinetics, Uncompensated resistance | Decrease scan rate, Check electrode connections, Use supporting electrolyte |
| Asymmetric peak currents | Chemical reactivity of redox species, Adsorption phenomena | Verify redox species stability, Clean electrode surface |
| High background current | Contaminated electrode, Electrolyte impurities | Repolish electrode, Use higher purity reagents |
| Non-reproducible peaks | Unstable reference electrode, Drifting open circuit potential | Condition reference electrode, Ensure stable temperature |
| No faradaic peaks | Incorrect potential window, Degraded redox species | Verify redox couple E⁰', Prepare fresh solutions |
Advanced Applications in Research
Beyond fundamental characterization, CV provides valuable insights for pharmaceutical and diagnostic applications. For biosensor development, CV can monitor biorecognition events through perturbations in impedance signals when target molecules bind to capture probes on electrode surfaces [40]. The technique enables optimization of signal-to-noise ratios through careful selection of redox probes and electrolyte compositions, facilitating transitions from expensive benchtop analyzers to affordable point-of-care devices [40]. Recent advances include non-triangular waveforms such as elliptic potential perturbations that may offer enhanced sensitivity for detecting multiple species with similar formal potentials [46]. Computational approaches combining density functional theory with experimental CV data further enhance understanding of redox mechanisms, particularly for systems involving coupled electron-proton transfer [41].
This application note provides comprehensive protocols for establishing cyclic voltammetry methodologies for redox reaction analysis. Proper execution of each step—from electrolyte preparation and electrode conditioning to instrument configuration and data interpretation—ensures reliable characterization of electrochemical systems. The systematic approach outlined enables researchers to obtain high-quality data for investigating redox mechanisms relevant to pharmaceutical development, diagnostic applications, and fundamental electrochemical studies. By adhering to these standardized procedures while understanding the theoretical principles underlying cyclic voltammetry, scientists can effectively utilize this powerful technique across diverse research applications.
Cyclic voltammetry (CV) is a powerful electroanalytical technique used to study redox reactions, providing significant insights into electrochemical properties, reaction mechanisms, and kinetics. For researchers in drug development, mastering CV is essential for investigating API redox behavior, stability, and interactions with biological targets like DNA. The reliability of CV data, however, depends critically on the meticulous optimization of experimental parameters. This Application Note details the roles of scan rate, analyte concentration, and pH, providing structured protocols to optimize these parameters for robust, reproducible results in pharmaceutical research.
The Scientist's Toolkit: Essential Research Reagents and Materials
The table below lists key reagents, materials, and equipment essential for conducting cyclic voltammetry experiments in a drug development context.
Table 1: Essential Research Reagents and Materials for CV in Drug Development
| Item | Specification / Typical Example | Primary Function in CV Experiment |
|---|---|---|
| Working Electrode | Glassy Carbon Electrode (GCE), Boron-Doped Diamond Electrode (BDDE) | Provides the surface for the redox reaction of the analyte. Material choice affects sensitivity and reproducibility. [26] [47] |
| Reference Electrode | Ag/AgCl (sat. KCl) | Provides a stable, known potential against which the working electrode potential is measured. [26] [47] |
| Counter Electrode | Platinum Wire | Completes the electrical circuit by facilitating current flow, preventing current limitation at the working electrode. [26] |
| Supporting Electrolyte | 0.1 M KCl, Phosphate Buffered Saline (PBS), Acetate Buffer | Carries current and minimizes resistive loss (iR drop). Determines the ionic strength and pH of the solution. [26] [47] [48] |
| Buffer System | Acetate Buffer (pH ~4.7), PBS (pH 7.4) | Maintains a stable pH environment, which is critical for proton-coupled electron transfer reactions. [47] [48] |
| Redox Analyte | Drug candidate (e.g., Ponatinib), Metal complex, Organic dye | The molecule of interest whose electrochemical redox behavior is under investigation. [26] [49] [47] |
| Solvent | Deionized Water, Ethanol | Dissolves the analyte, electrolyte, and other components. Must be electrochemically inert in the potential window of interest. [47] |
The Critical Triad: Parameters and Their Optimization
Scan Rate: Probing Kinetics and Reaction Control
The scan rate (ν) is the rate at which the applied potential is changed. It is a critical parameter for diagnosing whether a reaction is controlled by diffusion, adsorption, or involves coupled chemical steps.
Table 2: Scan Rate Effects on CV Data and Diagnostic Information
| Parameter / Relationship | Quantitative Relationship (Ideal System) | Diagnostic Information for Mechanism | ||
|---|---|---|---|---|
| Peak Current (ip) | ip ∝ ν1/2 (Randles-Ševčík equation) [50] [19] | Diffusion-controlled process: A plot of ip vs. ν1/2 is linear. Freely diffusing analyte. [50] | ||
| Peak Current (ip) | ip ∝ ν [50] | Surface-confined process: A plot of ip vs. ν is linear. Analyte is adsorbed onto the electrode surface. [50] | ||
| Peak Potential Separation (ΔEp) | ΔEp = | Epa - Epc | ≈ 59/n mV (at 25°C) for a reversible system [51] | Reversibility: ΔEp near this value indicates a reversible, fast electron transfer. Larger values suggest quasi-reversible or irreversible kinetics. [51] |
Experimental Protocol: Diagnosing Reaction Control via Scan Rate
- Objective: To determine whether the electrochemical reaction of a novel thiazolopyrimidine derivative is diffusion or adsorption controlled.
- Procedure:
- Prepare a solution of the drug candidate in an appropriate buffer (e.g., acetate buffer, pH 4.8) with supporting electrolyte. [49]
- Using a polished glassy carbon working electrode, Ag/AgCl reference electrode, and Pt counter electrode, record a series of CVs at scan rates from 10 mV/s to 1000 mV/s.
- For each voltammogram, measure the peak current (ip) for the reduction or oxidation peak of interest.
- Plot both ip vs. ν1/2 and ip vs. ν.
- Data Interpretation: A linear plot of ip vs. ν1/2 that passes through the origin confirms a diffusion-controlled process, as was determined for thiazolopyrimidine derivatives. [49] A linear plot of ip vs. ν indicates an adsorption-controlled process.
Analyte Concentration: Quantification and Binding Studies
The concentration of the electroactive species directly influences the peak current, enabling quantitative analysis. Furthermore, monitoring current changes upon addition of a binding partner (e.g., DNA) allows for the study of interaction strength and mode.
Table 3: Concentration Effects and Applications in Drug Development
| Application | Observed Change in CV | Key Outcome / Calculated Parameter |
|---|---|---|
| Quantitative Analysis | Peak current (ip) is directly proportional to analyte concentration. [50] | Enables construction of a calibration curve for determining unknown concentrations of an API. |
| Drug-DNA Interaction Studies | Decrease in drug peak current; shift in peak potential after DNA addition. [52] [47] | Indicates binding. The decrease is due to the reduced diffusion coefficient of the larger drug-DNA complex. [52] |
| Complexation Studies | Shift in redox peaks and change in currents upon addition of a complexing agent. [26] | Allows determination of complexation stability constants and Gibbs free energy of complexation (ΔG). [26] |
Experimental Protocol: Studying Drug-DNA Interactions via Concentration Titration
- Objective: To investigate the interaction between the anticancer drug Ponatinib and double-stranded DNA (dsDNA) and determine the resulting changes in the drug's electrochemical signal. [47]
- Procedure:
- Prepare a fixed concentration of Ponatinib (e.g., in PBS at physiological pH 7.4) and record a baseline CV or Square-Wave Voltammogram (SWV). [47]
- Sequentially add small, known aliquots of a concentrated dsDNA stock solution to the drug solution.
- After each addition, incubate the mixture for a fixed time (e.g., 5 minutes) and record the voltammogram.
- Measure the change in the peak current and/or peak potential of the drug after each DNA addition.
- Data Interpretation: A significant decrease in the voltammetric peak current of Ponatinib upon DNA addition confirms binding. [47] Analysis of the current decrease as a function of DNA concentration can be used to calculate the binding constant, providing a quantitative measure of interaction strength.
pH of the Medium: Influencing Thermodynamics and Mechanism
The pH of the electrolyte solution can dramatically alter the redox potential and mechanism, especially for organic molecules and APIs where electron transfer is often accompanied by proton transfer.
Experimental Protocol: Establishing the pH-Redox Potential Relationship for an API
- Objective: To characterize the electrochemical behavior of a Non-Steroidal Anti-Inflammatory Drug (NSAID) across a physiologically relevant pH range.
- Procedure:
- Prepare a series of solutions containing a fixed concentration of the NSAID in different buffers (e.g., Britton-Robinson buffer) covering a wide pH range (e.g., 3.0 to 9.0). [53]
- Record CVs for each solution under identical instrument settings (scan rate, sensitivity).
- For each pH, note the peak potential (Ep) and peak current (ip).
- Plot Ep vs. pH.
- Data Interpretation: A linear shift in Ep to more negative values with increasing pH indicates a proton-coupled electron transfer (PCET) reaction. The slope of the plot (dEp/dpH) can reveal the number of protons (m) involved per electron (n) transferred (slope ≈ -0.059 m/n V at 25°C). This information is crucial for understanding the drug's metabolic redox pathway and potential oxidative stress effects. [53]
Integrated Experimental Workflow
The following diagram illustrates the logical workflow for optimizing critical parameters in a cyclic voltammetry experiment, from initial setup to data interpretation and mechanism diagnosis.
Diagram 1: Workflow for optimizing a CV experiment, showing the iterative parameter optimization loop.
The rigorous optimization of scan rate, concentration, and pH is not merely a procedural step but a foundational practice for extracting meaningful electrochemical data. By systematically following the protocols outlined in this application note, researchers and drug development professionals can ensure their cyclic voltammetry experiments yield high-quality, reproducible results. Mastering these parameters unlocks the full potential of CV, enabling precise probing of redox mechanisms, reliable quantification of analytes, and detailed investigation of critical interactions, such as those between novel drug candidates and their biological targets. This systematic approach is indispensable for advancing pharmaceutical research and development.
Doxorubicin (DOX) is an anthracycline chemotherapeutic agent widely used in the treatment of various cancers, including breast cancer, leukemias, and lymphomas [54]. Despite its efficacy, the clinical use of DOX is limited by severe side effects, most notably dose-dependent cardiotoxicity, which can progress to irreversible cardiomyopathy and heart failure [54] [55]. The therapeutic monitoring of DOX is therefore crucial for optimizing dosage and minimizing toxic effects in patients.
Electrochemical methods, particularly voltammetry, have emerged as powerful alternatives to conventional analytical techniques like chromatography and spectrometry for drug monitoring [55]. These methods offer advantages of cost-effectiveness, rapid analysis, portability for point-of-care testing, and the ability to analyze complex biological samples with minimal pretreatment [54] [55]. This case study explores the application of voltammetric techniques for the detection of doxorubicin, framed within a broader thesis research on cyclic voltammetry for redox reaction analysis.
Electrochemical Properties of Doxorubicin
Doxorubicin contains a quinone-hydroquinone moiety in its anthraquinone aglycone structure, which confers its characteristic electrochemical redox activity [56] [57]. This functional group enables the molecule to undergo reversible electron transfer reactions, making it highly suitable for voltammetric analysis.
The electrochemical behavior of DOX is influenced by several factors:
- pH of the medium: The redox reaction involves proton transfer, with peak potentials shifting with changes in pH [57]. The electrochemical system can exhibit pseudo-reversible behavior in acidic media (e.g., acetate buffer, pH 3.5) [57].
- Interaction with metal ions: Doxorubicin can form complexes with Fe(III) ions, generating a distinct 1-electron reversible step at -0.494 V (vs. Ag|AgCl). This complex is more stable under aerobic conditions and may play a role in the drug's cardiotoxicity through the generation of reactive oxygen species [56].
- DNA binding: As an intercalating agent, DOX interacts with double-stranded DNA, which alters its electrochemical signals and can be exploited for sensitive detection [58] [59].
Table 1: Summary of Voltammetric Sensors for Doxorubicin Detection
| Electrode Modification | Technique | Linear Range | Limit of Detection | Sample Matrix | Reference |
|---|---|---|---|---|---|
| Pencil Graphite Electrode (PGE) | CV, DPV, LSV | Not specified | Good sensitivity | Pharmaceutical formulations | [60] |
| DNA-PolyPhTz/GCE | DPV | 10 pM - 0.2 mM | 5 pM | Artificial plasma, medications | [58] |
| MWCNTs/ZnO/SPCE | DPV | 0.007 - 150.0 µM | 0.002 µM | - | [61] |
| AuNPs/SPCE | DPV | 1 - 500 µg/mL | 0.3 µg/mL | Serum, pharmaceutical formulations | [54] |
| Paper-based Ag ink sensor | DPV | 10 - 1000 nM | 10 nM | Human plasma | [62] |
| CB-P5A-polyNR-MB/GCE | DPV | 10 nM - 0.1 mM | 10 nM | Synthetic blood plasma | [59] |
Advanced Sensor Platforms and Materials
Nanomaterial-Modified Electrodes
Carbon Nanotubes (CNTs) are extensively used in electrochemical sensors due to their exceptional conductivity, high surface area, and electrocatalytic properties [55]. MWCNTs/ZnO nanocomposite modified screen-printed carbon electrodes (SPCEs) demonstrate a significant synergistic effect, enhancing the redox reaction of DOX with an extremely low detection limit of 0.002 µM [61]. Similarly, oxidized MWCNTs on glassy carbon electrodes (OMWCNT/GCE) enable the simultaneous detection of DOX and dopamine by successfully separating their overlapped oxidation signals [55].
Metal and Metal Oxide Nanoparticles contribute unique properties to sensing platforms. Gold nanoparticle (AuNP)-modified in-house printed electrodes provide accurate quantification of DOX from novel pharmaceutical formulations and serum [54]. Fe₃O₄@Pt nanoparticles combined with MWCNTs on carbon paste electrodes enhance electrocatalytic performance for DOX detection in urine samples [55].
DNA-Based Sensors
Voltammetric DNA sensors exploit the intercalation of DOX into the DNA double helix for specific detection [58] [59]. These platforms typically consist of:
- Electrode substrate (e.g., glassy carbon electrode)
- Conductive nanomaterials (e.g., carbon black, MWCNTs)
- DNA immobilization layer (from fish sperm or salmon testes)
- Redox-active dyes (e.g., Neutral Red, Methylene Blue) as signal reporters
The interaction between DOX and DNA alters the electrochemical signals of the intercalated dyes, enabling quantitative detection of the drug [59]. DNA sensors can determine DOX in synthetic blood plasma with high sensitivity [59].
Innovative Substrates and Manufacturing
Paper-based sensors fabricated using pen-on-paper technology with highly conductive silver ink represent a promising approach for affordable, scalable biomedical diagnostics [62]. These sensors exhibit excellent thermal stability (up to 150°C), mechanical flexibility, and maintain functionality despite moisture absorption [62].
Screen-printed electrodes (SPEs) offer advantages of disposability, reproducibility, and integration with portable instrumentation, making them suitable for point-of-care monitoring of chemotherapeutic drugs [61] [54].
Experimental Protocols
This protocol outlines the fundamental procedure for studying doxorubicin at a pencil graphite electrode, suitable for initial electrochemical characterization.
Research Reagent Solutions:
- Doxorubicin standard solution: Prepare in appropriate solvent (e.g., water, buffer) at stock concentration of 1 mM
- Supporting electrolyte: 0.1 M phosphate buffer (pH 7.0) or acetate buffer (pH 3.5)
- Pencil graphite leads: Standard HB leads, 2B leads may offer different surface properties
Procedure:
- Electrode Preparation:
- Insert a pencil graphite lead into a suitable holder to establish electrical contact.
- Polish the electrode surface on fine emery paper or weighing paper to create a fresh, smooth surface.
- Rinse thoroughly with deionized water and dry gently.
Instrumental Setup:
- Configure a standard three-electrode system with:
- Working electrode: Prepared pencil graphite electrode
- Reference electrode: Ag/AgCl (3 M KCl)
- Counter electrode: Platinum wire
- Set up potentiostat with connections to all three electrodes.
- Configure a standard three-electrode system with:
Electrochemical Measurements:
- Place electrodes in supporting electrolyte solution containing various concentrations of DOX.
- Perform Cyclic Voltammetry (CV):
- Potential range: -0.8 to +0.8 V (vs. Ag/AgCl)
- Scan rate: 50-100 mV/s (optimize for reversible behavior)
- Perform Differential Pulse Voltammetry (DPV) for quantitative analysis:
- Potential range: 0 to +0.6 V (vs. Ag/AgCl)
- Pulse parameters: Amplitude 50 mV, pulse width 50 ms, scan rate 20 mV/s
- Record voltammograms for analysis.
Optimization Steps:
- Systematically vary pH (3-9) to identify optimal response.
- Evaluate effect of scan rate (10-500 mV/s) on peak currents.
- Study accumulation time/t potential if adsorption is observed.
Data Analysis:
- Measure peak currents (Ip) and potentials (Ep) from voltammograms.
- Plot calibration curve of Ip vs. DOX concentration.
- Calculate detection limit from signal-to-noise ratio (S/N=3).
This protocol describes the fabrication of a highly sensitive DNA sensor using electropolymerized films for doxorubicin determination in biological samples.
Research Reagent Solutions:
- Native DNA solution: Low-molecular DNA from salmon testes (<5% protein), dissolved in deionized water at 0.1-2.0 mg/mL
- Phenothiazine derivative solution: N-phenyl-3-(phenylimino)-3H-phenothiazin-7-amine (PhTz), 72 µM in acetone:0.4 M H₂SO₄ (1:1 v/v)
- Supporting electrolyte: Phosphate buffer (25 mM NaH₂PO₄ with 0.1 M Na₂SO₄, pH 5.0)
- Carbon black suspension: 1.3 mg/mL in 0.375% chitosan solution in 0.05 M HCl
Procedure:
- Electrode Modification:
- Polish glassy carbon electrode (GCE, 2 mm diameter) with alumina slurry (0.05 µm) and rinse thoroughly.
- Apply 1 µL carbon black suspension twice to GCE surface, drying at 60°C after each application.
- Apply 2 µL of pillar[5]arene solution (1×10⁻⁴ M in acetone) and dry in air.
Electropolymerization with DNA:
- Prepare electropolymerization solution containing 0.4 mM Neutral Red and DNA (0.5 mg/mL) in phosphate buffer (pH 6.0) with 0.1 M NaNO₃.
- Transfer modified electrode to polymerization solution.
- Perform cyclic voltammetry by scanning potential between -0.8 and +0.8 V (vs. Ag/AgCl) for 15-20 cycles at 50-100 mV/s.
- Transfer electrode to clean supporting electrolyte and record 10 CV cycles to remove unbound species.
Detection of Doxorubicin:
- Incubate DNA sensor in DOX solution (standard or sample) for 10-15 minutes.
- Transfer to electrochemical cell containing clean supporting electrolyte.
- Record DPV parameters:
- Potential range: -0.8 to 0 V (vs. Ag/AgCl)
- Amplitude: 50 mV
- Pulse width: 50 ms
- Scan rate: 20 mV/s
- Measure change in redox peak currents of polymerized dye.
Calibration and Quantification:
- Construct calibration curve using standard DOX solutions (10 nM - 0.1 mM).
- For biological samples, use standard addition method to account for matrix effects.
Diagram 1: DNA Sensor Fabrication Workflow (76 characters)
Data Analysis and Interpretation
Voltammetric Response Characteristics
The voltammetric detection of doxorubicin typically reveals distinct oxidation and reduction peaks corresponding to the quinone-hydroquinone redox couple. The peak potentials and currents provide quantitative and qualitative information about the drug.
Table 2: Electrochemical Signatures of Doxorubicin Under Different Conditions
| Electrode System | Medium/Conditions | Oxidation Peak (V) | Reduction Peak (V) | Notes | Reference |
|---|---|---|---|---|---|
| Bare PGE | pH 7.0 | +0.34 | Not specified | - | [55] |
| GCE | Acetate buffer, pH 3.5 | Not specified | Not specified | Pseudo-reversible behavior | [57] |
| Fe(III)-DOX complex | Pyrophosphate buffer, pH 9 | -0.494 | -0.494 | 1-electron reversible step | [56] |
| DNA-polyPhTz/GCE | Phosphate buffer | +0.5 | Not specified | DPV measurement | [58] |
Analytical Performance Comparison
The sensitivity and detection limits of DOX sensors vary significantly based on the electrode modification strategy and detection technique. Advanced nanomaterials and DNA-based sensors generally provide superior performance compared to unmodified electrodes.
Diagram 2: Sensor Design and Response Flow (65 characters)
Applications in Pharmaceutical and Clinical Analysis
Voltammetric sensors for DOX have been successfully applied to various real-world scenarios:
Pharmaceutical Formulation Analysis
The developed sensors can quantify DOX in commercial medications (e.g., Doxorubicin-TEVA and Doxorubicin-LANS) with recovery rates of 90-95% [58] [54]. This application is valuable for quality control in pharmaceutical manufacturing and verification of drug potency.
Biological Fluid Monitoring
Sensors have demonstrated capability to detect DOX in complex biological matrices including human plasma, synthetic blood plasma, urine, and serum [54] [62] [59]. The use of modified electrodes with enhanced selectivity enables direct analysis with minimal sample pretreatment in some cases.
Drug Release Studies from Novel Formulations
Electrochemical sensors provide a valuable tool for characterizing DOX-loaded nanocarriers (liposomes, niosomes, polymeric nanoparticles) during formulation development [54]. They enable rapid monitoring of drug loading efficiency and release profiles, facilitating optimization of novel drug delivery systems with improved safety profiles.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Research Reagent Solutions for Voltammetric Doxorubicin Detection
| Reagent/Material | Function/Purpose | Example Specifications |
|---|---|---|
| Doxorubicin hydrochloride | Primary analyte | Purity ≥ 95%, prepare stock solutions in water or buffer [54] |
| Pencil graphite leads | Economical electrode material | HB or 2B grade, suitable for disposable electrodes [60] |
| Multi-walled carbon nanotubes (MWCNTs) | Electrode nanomodifier | Enhance surface area, electron transfer; functionalized or oxidized forms available [61] [55] |
| Metal nanoparticles (Au, Ag, Pt) | Electrode nanomodifier | Catalytic activity, signal amplification; spherical, 10-50 nm diameter [54] |
| Native DNA | Biorecognition element | Salmon testes or fish sperm DNA, for DNA-based sensors [58] [59] |
| Phenothiazine dyes | Electropolymerization monomers | Neutral Red, Methylene Blue; form electroactive polymer films [59] |
| Supporting electrolytes | Provide ionic conductivity | Phosphate buffer (pH 5-8), acetate buffer (pH 3.5) [57] [59] |
| Chitosan | Biopolymer for immobilization | 0.375% solution in 0.05 M HCl; facilitates layer-by-layer assembly [59] |
Fast-Scan Cyclic Voltammetry (FSCV) for Real-Time Neurotransmitter Detection
Fast-scan cyclic voltammetry (FSCV) is an advanced electrochemical technique that has revolutionized the real-time detection of neurotransmitters in biological systems. Unlike traditional cyclic voltammetry with scan rates around 100 mV/s, FSCV operates at dramatically higher scan rates—typically 400-1000 V/s—enabling the acquisition of complete voltammograms within milliseconds [63] [64]. This exceptional temporal resolution allows researchers to monitor neurochemical dynamics at a biologically relevant time scale, capturing subsecond fluctuations in neurotransmitter concentrations that underlie fundamental brain functions [63] [65]. The technique's development and popularization by Millar and Wightman in the 1980s marked a significant milestone in analytical neurochemistry, providing unprecedented access to chemical signaling in the living brain [63] [65].
The core principle of FSCV involves applying a triangular waveform to a carbon-fiber microelectrode (CFME) immersed in the biological environment, typically scanning from a holding potential to a switching potential and back at high frequency [63]. When the electrode potential reaches the oxidation potential of a neurotransmitter, electron transfer occurs, generating a Faradaic current proportional to the analyte concentration [66]. The resulting cyclic voltammogram provides a distinctive electrochemical signature that aids in identifying the detected substance [64]. A critical aspect of FSCV is the background subtraction process, where the large capacitive charging currents are subtracted from the total current to reveal the Faradaic signal of interest [63]. This differential measurement approach enables exquisite sensitivity with detection limits for dopamine in the low nanomolar range (approximately 10 nM), making it suitable for monitoring subtle neurotransmitter fluctuations in extracellular space [67] [64].
Table 1: Comparative Analysis of Electrochemical Techniques for Neurotransmitter Detection
| Technique | Sensitivity | Selectivity | Temporal Resolution | Key Applications |
|---|---|---|---|---|
| FSCV | High (~10 nM LOD for dopamine) | Highest (CV shape identifies molecules) | High (100 ms) | Transient neurotransmitter release, uptake kinetics, behavioral studies |
| Amperometry | Low (25-100 nM LOD) | Low (detects all oxidizable compounds) | Highest (<1 ms) | Single exocytosis events, vesicular release |
| Chronoamperometry | Moderate | Moderate | Low (~1 s) | Pharmacological studies, basic oxidation measurements |
| Microdialysis | High (pM-nM) | High (with HPLC separation) | Very low (minutes) | Basal level measurements, metabolic profiling |
Experimental Setup and Instrumentation
Core System Components
The standard FSCV setup requires several integrated components to achieve optimal performance for neurotransmitter detection. At the heart of the system is the potentiostat, which applies the precise voltage waveforms and measures the resulting currents with nanoamp sensitivity [68]. Modern FSCV systems often incorporate digital circuits with ohmic drop compensation to minimize distortion at high scan rates, enabling accurate measurements even at rates exceeding 1000 V/s [68]. The working electrode is typically a carbon-fiber microelectrode (CFME) with diameters around 5-7 μm, fabricated by sealing a single carbon fiber in a glass capillary and cutting it to expose a clean disk surface [67] [69]. The reference electrode is usually a Ag/AgCl type, providing a stable potential reference in biological environments, while a auxiliary electrode completes the three-electrode system for in vitro applications, though two-electrode systems are often sufficient for in vivo measurements due to low currents [63] [67].
A critical advancement in FSCV instrumentation addresses the challenge of ohmic drop (iRu) compensation, which becomes significant at high scan rates due to solution resistance. Recent developments include digital circuits that precisely measure solution resistance online using impedance chips (e.g., AD5933) before implementing automatic positive feedback compensation through digital potentiometers [68]. This approach enables accurate voltammetric analysis at scan rates up to 1600 V/s without signal oscillation issues that plagued earlier compensation methods [68]. The system is controlled by specialized software that generates the waveform paradigms, collects the current data, and processes the signals through background subtraction and digital filtering algorithms to extract the Faradaic components [67] [70].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 2: Essential Research Reagents and Materials for FSCV
| Item | Specification | Function/Application |
|---|---|---|
| Carbon Fibers | 5-7 μm diameter, polyacrylonitrile-based (e.g., T650) | Working electrode material providing adsorption sites for neurotransmitters [69] |
| Borosilicate Glass Capillaries | 0.4 mm ID, 0.6 mm OD | Electrode insulation and structural support during fabrication [69] |
| Epoxy Resin | Spurr Epoxy or similar | Sealing the carbon fiber in the glass capillary [69] |
| Ag/AgCl Reference Electrode | Miniaturized for in vivo use | Stable potential reference in biological systems [63] |
| Artificial Cerebrospinal Fluid (aCSF) | Standard ionic composition (NaCl, KCl, CaCl2, MgCl2, HEPES) | In vitro calibration and brain slice experiments [69] |
| Neurotransmitter Standards | Dopamine HCl, serotonin HCl, adenosine, etc. | Electrode calibration and experimental controls [67] |
| Nafion Perfluorinated Polymer | 5% solution in aliphatic alcohols | Electrode coating to enhance selectivity and resist fouling [67] |
| Carbon Nanomaterials | Carbon nanotubes, graphene | Electrode modifications to enhance sensitivity and trapping molecules [63] [67] |
Core Methodologies and Protocols
Carbon-Fiber Microelectrode Fabrication Protocol
The fabrication of high-quality carbon-fiber microelectrodes is fundamental to successful FSCV experiments. The process begins with threading a single carbon fiber (7 μm diameter) into a borosilicate glass capillary (0.4 mm ID, 0.6 mm OD) [69]. The capillary is then pulled to a fine tip using a vertical pipette puller, creating a sealed glass sheath around the fiber. The tip is carefully sealed with epoxy resin to insulate the electrode and prevent solution leakage into the capillary [69]. The sealed electrode is cured at elevated temperature (typically 60°C for 24 hours) to ensure complete epoxy polymerization. Finally, the carbon fiber is trimmed approximately 100-400 μm from the glass seal using a surgical blade under microscopic guidance to create a clean, disk-shaped electrode surface [69]. Electrodes should be inspected under high magnification to ensure proper seal integrity and fiber alignment before use.
For enhanced performance and selectivity, CFMEs often undergo electrochemical pretreatment through the application of extended voltage waveforms. A common protocol involves immersing the electrode in phosphate-buffered saline and applying a triangle wave from -0.6 V to +1.4 V at 400 V/s for 1-2 hours [69]. This pretreatment creates oxygen-containing functional groups on the carbon surface that enhance cation adsorption and improve electron transfer kinetics [67]. For specific applications, electrodes may be modified with Nafion coatings (5% solution) to exclude anions like ascorbate and DOPAC, or with carbon nanomaterials such as carbon nanotubes to increase surface area and enhance sensitivity through analyte trapping mechanisms [63] [67].
Standard FSCV Measurement Protocol for Dopamine Detection
The following protocol details the optimized procedure for dopamine detection using FSCV, which can be adapted for other neurotransmitters with appropriate waveform modifications:
Electrode Conditioning: Before initial use, condition new CFMEs by applying the dopamine waveform (-0.4 V to +1.3 V, 400 V/s, 60 Hz) for 15-30 minutes in aCSF to stabilize the background current [63] [69].
System Calibration: Place the CFME in a flow injection system with continuous aCSF flow (1-2 mL/min). Apply the standard dopamine waveform (-0.4 V holding potential, +1.3 V switching potential, 400 V/s scan rate, 10 Hz repetition rate) and record the background current for 1-2 minutes until stable [63] [67].
Background Collection: Collect a stable background voltammogram (typically the average of 5-10 scans) for subsequent subtraction from experimental data.
Dopamine Detection: For in vitro measurements, inject dopamine standards (10 nM - 10 μM) in randomized order using a valve injection system with contact times of 1-5 seconds. For each injection, record the FSCV data, subtracting the background to obtain the Faradaic current [63] [66].
Data Acquisition: Collect current data at the peak oxidation potential for dopamine (~+0.6 V to +0.7 V) to create concentration-time traces for quantitative analysis. Simultaneously record full voltammograms every 100 ms for chemical identification [63] [66].
Post-experiment Calibration: After in vivo recordings or at the end of in vitro experiments, recalibrate the electrode to account for any sensitivity changes due to fouling or surface modification [67].
FSCV Waveform Optimization for Different Neurotransmitters
The applied voltage waveform is a critical parameter that determines FSCV sensitivity and selectivity for specific neurotransmitters. While the "dopamine waveform" (-0.4 V to +1.3 V, 400 V/s) serves as a standard starting point, optimized waveforms have been developed for various neurochemicals [67] [70]:
Serotonin Detection: Use an extended negative holding potential (-0.6 V to +1.4 V, 1000 V/s) to enhance sensitivity and reduce fouling by shifting oxidation products to more negative potentials [67].
Adenosine Detection: Apply a "N-shaped" waveform with a scan rate of 400 V/s from -0.6 V to +1.6 V and back to -0.6 V, followed by a step to +1.0 V before returning to the holding potential. This complex waveform helps distinguish adenosine from similar electroactive compounds [71].
Melatonin Detection: Implement a waveform scanning from -0.4 V to +1.4 V at 400 V/s with a holding potential of -0.4 V, which minimizes electrode fouling from oxidation byproducts while maintaining sensitivity [69].
Hydrogen Peroxide Detection: Utilize a restricted voltage range (-0.6 V to +1.2 V, 400 V/s) to avoid oxygen evolution interference while maintaining sufficient driving force for H2O2 oxidation [67].
Table 3: Optimized FSCV Waveform Parameters for Different Neurochemicals
| Analyte | Holding Potential (V) | Switching Potential (V) | Scan Rate (V/s) | Key Modifications |
|---|---|---|---|---|
| Dopamine | -0.4 | +1.3 | 400 | Standard waveform; enhances cation adsorption [63] |
| Serotonin | -0.6 | +1.4 | 1000 | Extended negative potential reduces fouling [67] |
| Adenosine | -0.6 | +1.6 | 400 | N-shaped waveform improves selectivity [71] |
| Melatonin | -0.4 | +1.4 | 400 | Minimizes oxidation byproduct deposition [69] |
| Histamine | -0.5 | +1.5 | 600 | Intermediate parameters balance sensitivity/selectivity [67] |
| Norepinephrine | -0.4 | +1.3 | 400 | Similar to dopamine but different CV signature [64] |
| Hydrogen Peroxide | -0.6 | +1.2 | 400 | Restricted range avoids oxygen interference [67] |
Data Analysis and Interpretation
Signal Processing and Chemical Identification
FSCV generates substantial datasets, with a typical one-hour experiment producing 36,000 individual cyclic voltammograms when collected at 10 Hz [67]. The initial data processing step involves background subtraction, where the capacitive charging current is subtracted from each scan to isolate the Faradaic signal [63]. The resulting background-subtracted voltammograms are visualized using false color plots, which provide a bird's-eye view of current as a function of both applied potential and time, enabling rapid identification of neurochemical release events and their characteristic electrochemical signatures [67].
Chemical identification relies on analyzing the shape of the cyclic voltammogram, particularly the oxidation and reduction peak potentials and their relative currents [63] [64]. For example, dopamine exhibits oxidation at approximately +0.6 V to +0.7 V and reduction at -0.2 V to -0.3 V vs. Ag/AgCl, with the reduction peak typically smaller than the oxidation peak due to chemical rearrangement of the o-quinone product [63] [66]. To distinguish neurotransmitters with similar electrochemical properties, multivariate analysis techniques such as principal component regression (PCR) are employed [63] [65]. PCR utilizes training sets of known compounds to create mathematical models that can automatically identify and quantify specific analytes in complex biological mixtures, effectively separating dopamine signals from pH changes or other interfering species [63] [65].
Kinetic Analysis of Neurotransmitter Release and Uptake
FSCV coupled with electrical stimulation provides powerful insights into the kinetics of neurotransmitter release and reuptake [65]. Typical experiments involve applying brief, localized electrical stimuli (e.g., 1-2 s trains of biphasic pulses) to neuronal pathways while monitoring neurotransmitter transients in projection regions. The resulting data are analyzed using Michaelis-Menten kinetic models to extract key parameters: the concentration of neurotransmitter released per stimulation pulse ([DA]p) and the maximal uptake rate (Vmax) [65]. This approach has revealed critical adaptations in dopamine systems in models of drug addiction, Parkinson's disease, and schizophrenia [65].
For behavioral experiments with freely moving animals, FSCV recordings capture naturally occurring phasic neurotransmitter transients that correlate with specific behaviors or cognitive processes [65]. Analysis focuses on identifying these transients above the background noise, typically using automated algorithms based on signal-to-noise thresholds or machine learning approaches [65]. Recent advances include the application of machine learning methods to identify dopamine transients in FSCV recordings from the human brain, demonstrating the translational potential of this technique [65].
Applications in Neurochemical Research
Investigating Neurotransmitter Dynamics in Neuropsychiatric Disorders
FSCV has generated groundbreaking insights into the neurochemical underpinnings of various neuropsychiatric disorders by revealing alterations in dopamine signaling dynamics. In substance use disorders, FSCV studies have demonstrated that subsecond dopamine release events in the nucleus accumbens precede cocaine-seeking behavior in self-administering animals, suggesting a potential role in promoting drug-seeking motivation [65]. Furthermore, repeated cocaine exposure enhances cue-evoked phasic dopamine release, creating a neuroadaptation that may contribute to addiction vulnerability [65].
In Parkinson's disease models, FSCV has been instrumental in characterizing the progressive loss of dopamine release and alterations in uptake kinetics that accompany dopaminergic degeneration [65]. These studies have revealed compensatory mechanisms in remaining dopamine terminals, including increased release per terminal and modified uptake kinetics, which have important implications for understanding disease progression and treatment strategies [65]. For schizophrenia research, FSCV has helped identify aberrant dopamine signaling patterns that may contribute to positive symptoms, particularly by examining dopamine system responsivity to stimuli and pharmacological challenges [65].
Expanding Beyond Monoamines: Novel Applications
While initially developed for catecholamine detection, FSCV applications have expanded to encompass a growing range of neurochemicals. Adenosine detection with FSCV has revealed a previously unappreciated rapid signaling mode that occurs on a timescale of seconds rather than the traditionally studied minutes-to-hours modulation [71]. This rapid adenosine release is activity-dependent and modulates oxygen levels and evoked dopamine release, suggesting novel regulatory mechanisms in the brain [71]. Similarly, FSCV detection of melatonin in the brain has been recently demonstrated using electrochemically pre-activated carbon-fiber electrodes, achieving unprecedented sensitivity (28.1 nA/μM) and low detection limits (20.2 ± 4.8 nM) [69]. This advancement enables real-time monitoring of this important neurohormone in various brain functions and potential therapeutic applications.
Other novel applications include FSCV measurements of neuropeptides using optimized waveforms and modified electrodes, hydrogen peroxide dynamics associated with oxidative stress and signaling, and guanosine in purinergic signaling pathways [67] [70]. The continuous development of new waveforms and electrode materials continues to expand the neurochemical landscape accessible to FSCV investigation, opening new frontiers in understanding brain chemistry.
Electrochemical Assessment of Antioxidant Activity in Natural Compounds
The electrochemical assessment of antioxidant activity leverages the fundamental principle that antioxidants are electron donors. Cyclic voltammetry (CV) has emerged as a powerful, rapid, and inexpensive technique to characterize the redox behavior and antioxidant potential of natural compounds by measuring their oxidation/reduction potentials [72] [73]. When an antioxidant donates an electron at the electrode surface, it generates a current; the potential at which this oxidation occurs is directly related to the compound's antioxidant strength, while the current magnitude relates to its concentration or the total capacity [74] [28]. This provides a direct measurement of a sample's reducing power, which is often well-correlated with its ability to scavenge free radicals in biological systems [73]. Unlike traditional spectrophotometric assays (e.g., DPPH, ABTS), which can be hampered by colored samples, require specific reagents, and measure only a particular mechanism, CV can offer a more holistic view of the total antioxidant capacity and the redox mechanisms involved [75] [76] [77]. This methodology is particularly valuable for screening complex mixtures like plant extracts, foods, and dietary supplements, where synergistic interactions between multiple antioxidant compounds can occur [78] [73].
The following diagram illustrates the core operational workflow of a Cyclic Voltammetry experiment for antioxidant assessment.
Optimized Experimental Protocols
Protocol 1: CV Analysis of Phenolic Compounds and Amino Acids
This protocol is adapted from a study investigating the synergistic antioxidant effects between phenolic compounds and amino acids [72] [78].
- Equipment and Reagents: Potentiostat (e.g., AUTOLAB PGSTAT 302 N) with three-electrode system: glassy carbon working electrode, platinum auxiliary electrode, and Ag/AgCl reference electrode. Standard compounds: gallic acid, catechin, caffeic acid, quercetin, cysteine, tryptophan, etc. Acetate buffer (0.1 M, pH 3.0) and phosphate buffer (0.1 M, pH 7.0). Methanol and purified water.
- Electrode Preparation: Prior to each measurement, polish the glassy carbon working electrode in an alumina/water suspension on a polishing cloth. Rinse thoroughly with purified water and then sonicate for 2 minutes to remove any adsorbed particles [72] [79].
- Sample Preparation:
- Individual Solutions: Prepare stock solutions of phenolic compounds at 0.5-1.0 g/L and amino acids at 1.0-2.0 g/L in suitable solvents (e.g., methanol or buffer) [78].
- Binary/Mixed Solutions: Prepare mixtures with phenolic compounds at 1.0 g/L and amino acids at 2.0 g/L to study synergistic effects [72].
- For measurement, dilute 1 mL of the model solution with 25 mL of the appropriate buffer [72].
- Measurement Conditions:
- For phenolic compounds, use acetate buffer (pH 3.0) and a scan range from 0.0 to 1.0 V [72] [78].
- For amino acids and mixed solutions, use phosphate buffer (pH 7.0) and a scan range from 0.0 to 2.0 V [72] [78].
- Set the scan rate to 100 mV/s. Deaerate the solution in the electrochemical cell with nitrogen (N₂) for 10 minutes before initiating the scan to remove dissolved oxygen. Maintain the temperature at 25.0 ± 0.5 °C [72].
- Data Analysis: Record the cyclic voltammogram. Identify the anodic peak potential (Epa), which indicates the antioxidant strength (a lower Epa suggests a stronger antioxidant). Determine the anodic peak current (Ipa), which is proportional to the concentration of oxidizable compounds. The onset potential (Eon) can also be used to evaluate synergistic effects [72].
Protocol 2: CV Analysis of Crude Plant Extracts
This protocol is suitable for screening the antioxidant potential of complex plant matrices [28] [73].
- Equipment and Reagents: Potentiostat with standard three-electrode system (Glassy Carbon, SCE/AgAgCl, Pt). Methanol or ethanol for extraction. Phosphate buffer (0.2 M, pH 7.0) as supporting electrolyte.
- Sample Preparation: Extract dried and powdered plant material (e.g., 100 g) with methanol (3 x 300 mL) using maceration at room temperature. Filter and concentrate the combined extracts under reduced pressure. For CV analysis, dissolve the concentrated extract in methanol at a concentration of 0.5 g/100 mL. Due to low water solubility, a mixture of acetonitrile and buffer may be used [73].
- Measurement Conditions: Use phosphate buffer (0.2 M, pH 7.0) as the supporting electrolyte. A scan rate of 100 mV/s is typical, but a range from 10 to 400 mV/s can be used to study the effect of scan rate. The potential scan range should typically be from 0.0 to +1.0 V (vs. Ag/AgCl) [73].
- Data Analysis: Analyze the voltammogram for the number of anodic peaks, which represent different classes of oxidizable compounds. A low first oxidation potential (Epa < +0.4 V) indicates high antioxidant capacity, as the compounds are more easily oxidized. A high peak current (Ipa) suggests a high content of redox-active compounds [28] [73].
Protocol 3: Correlation with Lipid Peroxidation Inhibition (TBARS Assay)
To validate the electrochemical data with a biologically relevant antioxidant assay, the TBARS method can be employed [74].
- TBARS Assay Procedure: Prepare a tissue homogenate (e.g., from rat liver) and induce lipid peroxidation using an appropriate pro-oxidant system (e.g., Fe²⁺/ascorbate). Incubate the homogenate with and without the antioxidant sample (e.g., plant extract). After incubation, add thiobarbituric acid (TBA) reagent and heat to form the pink TBA-malondialdehyde (MDA) complex. Measure the absorbance of this complex at 532 nm [74].
- Data Correlation: The percentage of lipid peroxidation inhibition is calculated relative to a control. Perform simple regression analysis between the CV parameters (such as anodic peak current or peak area) and the percentage of inhibition obtained from the TBARS assay. A high correlation coefficient confirms that the CV response is a good indicator of the sample's bioactivity in inhibiting lipid oxidation [74].
Quantitative Data and Electrochemical Parameters
The following tables summarize key electrochemical parameters and optimization data from recent studies.
Table 1: Key Electrochemical Parameters Obtained from Cyclic Voltammetry and Their Significance in Antioxidant Assessment.
| Parameter | Description | Significance in Antioxidant Assessment |
|---|---|---|
| Anodic Peak Potential (Epa) | Potential at the maximum oxidation current. | Indicates antioxidant strength; a lower Epa signifies a higher antioxidant potential as it is more easily oxidized [72] [73]. |
| Anodic Peak Current (Ipa) | Maximum current of the oxidation peak. | Proportional to the concentration of oxidizable (antioxidant) compounds in the sample [74] [73]. |
| Onset Potential (Eon) | Potential where oxidation current begins to increase significantly. | Used to evaluate the thermodynamic tendency to donate electrons; useful for studying synergistic effects [72]. |
| Peak Area | Area under the oxidation peak. | Represents the total charge transferred, correlating with the total antioxidant capacity [74]. |
Table 2: Optimized Electrochemical Conditions for Different Compound Classes as Determined by Experimental Optimization [72] [78].
| Compound Class | Optimal pH | Optimal Concentration | Scan Range | Functional Insights |
|---|---|---|---|---|
| Phenolic Compounds (e.g., Gallic acid, Catechin) | 3.0 (Acetate buffer) | 0.5 - 1.0 g/L | 0.0 to +1.0 V | Lower Epa in binary/mixtures indicates synergistic antioxidant enhancement [72] [78]. |
| Amino Acids (e.g., Tryptophan, Cysteine) | 7.0 (Phosphate buffer) | 1.0 - 2.0 g/L | 0.0 to +2.0 V | Electrochemical activity is pH-dependent and related to specific functional groups (e.g., -SH, -NH₂) [72]. |
| Mixed Phenol-Amino Acid Solutions | 7.0 (Phosphate buffer) | Phenol: 1.0 g/LAmino Acid: 2.0 g/L | 0.0 to +2.0 V | Demonstrated synergistic effect, with Epa values lower than individual solutions [72]. |
| Crude Plant Extracts | 7.0 (Phosphate buffer) | Variable (e.g., 0.5 g/100 mL) | 0.0 to +1.0 V | Multiple anodic peaks indicate presence of various antioxidant compounds with different redox potentials [73]. |
The Researcher's Toolkit: Essential Reagents and Materials
Table 3: Essential Research Reagent Solutions and Materials for Electrochemical Antioxidant Assessment.
| Item | Typical Specification / Example | Function / Purpose |
|---|---|---|
| Potentiostat/Galvanostat | AUTOLAB PGSTAT series, CHI 660B | Instrument for applying potential and measuring current; the core of the CV setup [72] [79]. |
| Glassy Carbon Electrode (GCE) | 3 mm diameter, polished with alumina | The standard working electrode where the oxidation of antioxidants occurs [72] [80]. |
| Reference Electrode | Ag/AgCl (3 M KCl) | Provides a stable and known reference potential for the working electrode [72] [73]. |
| Counter Electrode | Platinum wire or coil | Completes the electrical circuit by conducting current from the working electrode [72] [73]. |
| Buffer Solutions | Acetate buffer (pH 3.0), Phosphate buffer (pH 7.0) | Serves as the supporting electrolyte, controlling pH and ionic strength of the solution [72] [78]. |
| Antioxidant Standards | Gallic acid, Quercetin, Ascorbic acid, Trolox | Used for calibration, method validation, and as reference compounds [72] [79]. |
| Polishing Supplies | Alumina slurry (1.0, 0.3, and 0.05 µm) | For polishing the GCE surface to ensure reproducibility and a clean, active surface [72] [79]. |
Application Workflow and Data Interpretation
The practical application of CV for antioxidant assessment involves a logical sequence from sample preparation to data interpretation, which can be integrated with other assays for validation. The following diagram outlines this workflow.
The interpretation of cyclic voltammetry data provides multi-faceted insights into a sample's antioxidant profile. The anodic peak potential (Epa) is a crucial parameter for ranking antioxidant strength. For instance, plant extracts with an Epa around +0.3 V (vs. Ag/AgCl) are considered to have strong reducing potential, whereas an Epa at +0.5 V or higher indicates weaker antioxidant activity or potential pro-oxidant behavior [73]. The anodic peak current (Ipa) provides a quantitative measure related to the total concentration of oxidizable compounds. This has been shown to correlate well with the total phenolic content determined by conventional assays like Folin-Ciocalteu [74] [80]. Furthermore, CV is exceptionally powerful for identifying synergistic effects in mixtures. Research has demonstrated that binary mixtures of phenolic compounds or mixed phenol-amino acid solutions often exhibit a lower Epa (i.e., they are more easily oxidized) compared to the individual compound solutions, indicating an enhanced antioxidant potential through synergy [72]. Finally, for biological relevance, the electrochemical data should be correlated with established antioxidant or anti-peroxidation assays. A strong positive correlation between the voltammetric peak area and the inhibition of lipid peroxidation in the TBARS assay has been reported, validating CV as a predictive tool for bioactivity [74].
Cyclic Voltammetry (CV) is a cornerstone electrochemical technique, fundamental for characterizing redox behavior by measuring the current resulting from a cyclically swept potential applied to a working electrode [81] [18]. While it excels at providing macroscopic, averaged data on electron transfer processes, diffusion coefficients, and reaction kinetics, its spatial resolution is inherently limited [30]. Understanding electrochemical activity at the nanoscale—where the influence of the Electric Double Layer (EDL) is pronounced and confinement effects dramatically alter reaction dynamics—requires innovative tools that transcend these classical limitations [30].
Opto-iontronic microscopy emerges as a revolutionary methodology that integrates advanced optical microscopy with nanoelectrode technology, enabling direct, label-free optical voltammetry within attoliter (10⁻¹⁸ L) volumes [30] [82]. This technique moves beyond passive observation, allowing for the real-time monitoring of ion concentration changes and redox reactions in nanoconfined environments, thus providing unprecedented mechanistic insights that are obscured in conventional macroscopic CV measurements [30]. This application note details the protocols and applications of this powerful technique, framing it within the ongoing evolution of redox reaction analysis.
Experimental Principles and Workflow
Opto-iontronic microscopy operates on the principle of detecting minute optical changes caused by ion concentration variations within a nanoconfined electrochemical cell during potential application [30] [82]. The core setup utilizes Total Internal Reflection (TIR) illumination to create an evanescent field that selectively probes the nanoscale region of interest, coupled with lock-in detection to achieve a high signal-to-noise ratio capable of detecting modulation ratios as low as 10⁻⁶ [30] [82] [83].
Table 1: Core Components of an Opto-iontronic Microscopy Setup
| Component Category | Specific Element | Function & Specification |
|---|---|---|
| Optical System | TIR Illumination (e.g., 640 nm laser) | Creates an evanescent field penetrating ~100-200 nm, selectively illuminating nanoholes or electrode tips near the interface [30] [83]. |
| High-NA Objective (e.g., 60x, 1.4 NA) | Used for both generating TIR and collecting scattered light [83]. | |
| Detection Paths | sCMOS camera for spatial mapping; Photodiode + Lock-in Amplifier for high-sensitivity, real-time measurement [30]. | |
| Electrochemical Cell | Nanohole Array Electrode | Cylindrical nanoholes (e.g., 75 nm diameter, 100 nm depth) in a Au/SiO₂ stack on glass, creating ~0.4 attoliter electroactive volumes [30] [82]. |
| Reference Electrode (e.g., Ag/AgCl) | Provides a stable potential reference [18]. | |
| Counter Electrode (e.g., Pt wire) | Completes the electrical circuit [18]. | |
| Electronic Control & Data Acquisition | Potentiostat | Precisely controls the cell potential (DC and AC components) [83]. |
| Lock-in Amplifier | Extracts the amplitude and phase of the weak optical signal synchronized to the AC potential modulation [30] [83]. | |
| Waveform Generator/DAQ Card | Generates potential waveforms and synchronizes all instruments [83]. |
The following diagram illustrates the integrated workflow of the technique, from sample preparation to data acquisition and interpretation.
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful implementation of opto-iontronic microscopy relies on a specific set of materials and reagents, each serving a critical function in the nanoscale electrochemical and optical system.
Table 2: Essential Research Reagent Solutions and Materials
| Item Name | Function / Role | Specification / Notes |
|---|---|---|
| Nanohole Array Electrode | Creates nanoconfined electrochemical environment. | Fabricated via Focused Ion Beam (FIB) milling; 100 nm Au layer on glass; nanoholes ~75 nm diameter, 100 nm depth [30] [82]. |
| Redox-Active Species | Model system to study electron transfer kinetics. | 1,1'-Ferrocenedimethanol (Fc(MeOH)₂); well-characterized, reversible redox couple; typically used at 0.1-10 mM [30] [82]. |
| Supporting Electrolyte | Provides ionic conductivity; minimizes solution resistance. | Potassium Chloride (KCl); commonly used at 100 mM to 1.5 M [30] [82]. |
| Reference Electrode | Maintains a stable and known electrochemical potential. | Ag/AgCl in 3M KCl is a standard choice [18]. |
| Total Internal Reflection (TIR) Buffer | Matches refractive index for optimal TIR illumination. | Standard aqueous buffers (e.g., phosphate buffer) can be used, provided they are compatible with the electrolyte and redox species [30]. |
Detailed Experimental Protocols
Protocol 1: Nanohole Electrode Fabrication and Characterization
Objective: To fabricate and validate a nanoelectrode array suitable for attoliter-volume redox imaging [30] [82].
- Substrate Preparation: Begin with a clean glass coverslip. Deposit a 50 nm layer of SiO₂ as an insulating layer, followed by a 100 nm layer of gold via physical vapor deposition (e.g., sputtering).
- Focused Ion Beam (FIB) Milling: Mount the substrate in an FIB instrument. Using a focused Ga⁺ ion beam, drill an array of cylindrical nanoholes through the Au/SiO₂ stack. A typical pattern is a square lattice with a 6 µm pitch to prevent optical cross-talk.
- Target Geometry: Aim for nanoholes with a diameter of approximately 75 nm and a depth of 100 nm, resulting in an electroactive volume of roughly 0.4 attoliters.
- Quality Control: Characterize the fabricated nanohole array using Scanning Electron Microscopy (SEM) to confirm the diameter, circularity, and placement of the nanoholes [30].
Protocol 2: Optical Configuration for EDL Modulation Microscopy
Objective: To align the TIR illumination and high-sensitivity detection path [30] [83].
- Laser Alignment: Couple a 640 nm laser diode into the microscope path. Focus the beam at the edge of the back focal plane (BFP) of a high numerical aperture (NA ≥ 1.4) oil-immersion objective to achieve TIR at the glass-electrolyte interface.
- Sample Positioning: Place the nanohole electrode sample on the stage with the electrode surface facing the objective. Introduce the electrolyte solution.
- Detection Path Setup:
- Imaging Path: Direct the scattered light to an sCMOS camera. Fine-focus to obtain a clear, stable image of the nanohole array.
- High-Sensitivity Path: Direct a portion of the scattered light to a photodetector. Connect the photodetector output to a lock-in amplifier.
- Lock-in Amplifier Setup: Set the lock-in amplifier to the same frequency as the applied AC potential modulation (e.g., 75 Hz). Adjust the phase and time constant for optimal signal-to-noise ratio.
Protocol 3: Alternating Current Voltammetry (ACV) with Lock-in Detection
Objective: To acquire correlated electrochemical and optical data during a redox reaction [30] [82] [83].
- Solution Preparation: Prepare an electrolyte solution containing 100 mM KCl as supporting electrolyte and 1 mM Fc(MeOH)₂ as the redox species. Degas if necessary.
- Electrochemical Cell Assembly: Assemble a three-electrode cell using the nanohole array as the working electrode, along with a reference electrode (e.g., Ag/AgCl) and a Pt counter electrode.
- Potential Waveform Application: Use the potentiostat to apply a composite potential waveform to the working electrode:
- DC Component: A slow triangular scan (e.g., 10-50 mV/s) from -0.2 V to 0.2 V vs. Ref.
- AC Component: A sinusoidal modulation (e.g., 50 mV amplitude, 75 Hz) superimposed on the DC ramp.
- Synchronized Data Recording:
- Record the traditional DC and AC electrochemical currents from the potentiostat.
- Simultaneously, record the amplitude (and phase) of the modulated optical scattering intensity from the lock-in amplifier, which corresponds to the local concentration changes of ionic species within the nanohole.
Data Interpretation and Mechanistic Insights
The key innovation of opto-iontronic microscopy is its ability to directly correlate optical signals with theoretical models to reveal nanoscale mechanisms. The experimental data is validated against a Poisson-Nernst-Planck-Butler-Volmer (PNP-BV) model, which calculates time-dependent ion concentration profiles [30] [82].
Table 3: Key Experimental Observations and Their Interpretation
| Observation | Experimental Condition | Theoretical Insight & Significance |
|---|---|---|
| Linear optical response vs. potential [82] | Pure KCl electrolyte (no redox species). | Signal is dominated by EDL (dis)charging; symmetric accumulation/depletion of K⁺ and Cl⁻ ions. Serves as a capacitive baseline. |
| Systematic deviation from linearity; drop in optical amplitude [30] [82] | Electrolyte with Fc(MeOH)₂ within its redox potential window. | PNP-BV model reveals large concentration oscillations (>50%) of Fc/Fc⁺ species, confirming optical contrast is primarily from redox-active species concentration. |
| Optical amplitude drop is concentration-dependent [82] | Varying Fc(MeOH)₂ concentration (0.1 - 10 mM). | Larger optical signal drop with higher concentration; validates quantitative sensing capability. |
| Frequency-dependent optical response [82] | Varying AC modulation frequency. | Response decreases at higher frequencies, governed by the RC time constant of the nanohole, informing on ion transport dynamics. |
The following diagram illustrates the fundamental signaling pathway of how an applied potential is transduced into a quantifiable optical signal via nanoscale electrochemical activity.
Application in Redox Reaction Analysis
Within the broader context of cyclic voltammetry research, opto-iontronic microscopy addresses a critical gap: the spatiotemporal monitoring of electrochemical processes at the nanoscale. While traditional CV provides bulk thermodynamic and kinetic parameters, this technique reveals the underlying ionic fluxes and concentration gradients that define these parameters in confined spaces [30]. It enables the direct observation of phenomena such as the coupling between EDL structure and redox kinetics, which is paramount for developing next-generation electrochemical devices including nano-sensors, advanced batteries, and electrocatalysts [30] [84]. The methodology's label-free nature and ultra-small detection volume also pave the way for future applications in monitoring nanocrystal growth and, potentially, single-molecule electrochemistry [30].
Troubleshooting CV Experiments: Solving Common Problems and Optimizing Data Quality
A General Troubleshooting Procedure for Equipment and Electrodes
Cyclic voltammetry (CV) is a powerful and versatile electrochemical technique used extensively to study redox processes, reaction mechanisms, and electron transfer kinetics [85]. Despite its widespread application in research and drug development, obtaining high-quality, reproducible voltammograms can be challenging due to equipment malfunctions and electrode issues. This application note details a systematic troubleshooting procedure to help researchers identify and resolve common hardware and electrode problems, ensuring the integrity of electrochemical data for redox reaction analysis.
Theoretical Background: The CV Setup
A standard cyclic voltammetry experiment requires a potentiostat to control the potential and measure the current in a three-electrode cell [85] [86]. The working electrode (WE) is where the redox reaction of interest occurs. The reference electrode (RE) (e.g., Ag/AgCl) provides a stable, known potential against which the WE is controlled. The counter electrode (CE) completes the electrical circuit. The experimental solution consists of a solvent, a high concentration of electrolyte (0.05–0.5 M) to minimize solution resistance, and the analyte at a lower concentration (typically 1–10 mM) [86].
Troubleshooting Methodology
When faced with an unusual cyclic voltammogram, a systematic approach is crucial. The following procedure, adapted from established methodologies [87], isolates problems with the potentiostat, cables, and electrodes.
The diagram below outlines the logical flow for diagnosing common equipment and electrode issues.
Detailed Diagnostic Steps
Step 1: Potentiostat and Cable Verification
- Procedure: Disconnect the electrochemical cell. Connect a 10 kΩ resistor between the working electrode cable and the combined reference and counter electrode cables. Run a potential scan from +0.5 V to -0.5 V [87].
- Expected Result: A straight, ohmic current-response line following V = IR [87].
- Interpretation: If this response is not observed, the issue likely lies with the potentiostat or the connecting cables. If the response is correct, the problem is likely in the cell or electrodes.
Step 2: Test Chip Scan (if available)
- Procedure: Use the manufacturer's test chip (e.g., the Ossila Test Cell Chip). Connect the potentiostat to a known test resistor on the chip (e.g., WE4) and perform a single cycle scan from 0 to 1 V at 100 mV/s [87].
- Expected Result: A straight line from 0 to 1 μA [87].
- Interpretation: A failed test confirms a potentiostat or cable issue.
Step 3: Reference Electrode Bypass
- Procedure: Set up the cell normally, but connect the reference electrode cable to the counter electrode (in addition to the CE cable). Run a linear sweep voltammetry experiment with your analyte present [87].
- Expected Result: A voltammogram that appears standard but is shifted in potential and slightly distorted due to increased uncompensated resistance [87].
- Interpretation: If a standard voltammogram is obtained, the original reference electrode is faulty. If the signal remains problematic, the issue likely lies with the working or counter electrodes.
Step 4: Working Electrode Inspection and Cleaning
- Procedure: If the previous steps point to an electrode issue, polish the working electrode with 0.05 μm alumina slurry and wash it thoroughly. For Pt electrodes, further clean by cycling in 1 M H2SO4 between the potentials for H₂ and O₂ evolution [87]. Also, check for poor internal contacts or broken seals in the electrode body [87].
- Interpretation: This addresses common problems like adsorbed contaminants or poor electrical contacts that lead to high resistance, noise, or sloping baselines.
Common Problems & Solutions
Based on the diagnostic procedure, the following table summarizes common observable issues, their likely causes, and recommended solutions.
Table 1: Common Cyclic Voltammetry Issues and Solutions
| Observed Problem | Potential Cause | Recommended Solution |
|---|---|---|
| Voltage Compliance Error [87] | Quasi-reference electrode touching WE; CE disconnected or out of solution. | Ensure all electrodes are properly submerged and not touching; check cable connections. |
| Current Compliance Error / Shutdown [87] | Working and Counter electrodes are touching, causing a short circuit. | Separate the electrodes and ensure they are properly positioned in the cell. |
| Flatlining Signal [88] | Current range setting is too low for the actual current, causing clipping. | Increase the current range setting on the potentiostat (e.g., to 1000 µA). |
| Unusual Voltammogram / Different on Repeated Cycles [87] | Reference electrode not in electrical contact (blocked frit/air bubbles). | Check/clean reference electrode frit; use it as a quasi-reference to verify; ensure no contact with CE. |
| Very Small, Noisy Current [87] | Working electrode is not properly connected to the cell. | Check WE connection and cable; ensure electrode is fully submerged. |
| Non-Flat Baseline [87] | Problems with the working electrode or unknown interfacial processes. | Polish and clean the WE; check for electrode faults. |
| Large Reproducible Hysteresis in Baseline [87] | Charging currents at the electrode-solution interface. | Reduce scan rate, increase analyte concentration, or use a smaller WE. |
Essential Research Reagents and Materials
The table below lists key materials required for a robust cyclic voltammetry experiment and for executing the troubleshooting protocol.
Table 2: Key Research Reagent Solutions and Materials
| Item | Function / Purpose |
|---|---|
| Supporting Electrolyte (e.g., KCl, NBu₄PF₆) [86] [28] | Minimizes solution resistance and carries current, without reacting in the potential window of interest. |
| High-Purity Solvent (e.g., Acetonitrile, Ethanol) [28] [89] | Dissolves analyte and electrolyte; must be electrochemically inert in the scanned potential range. |
| Standard Redox Probe (e.g., Ferrocene) [28] [89] | Used for system validation and referencing potentials, known for its reversible electrochemistry. |
| Alumina Polishing Slurry (0.05 μm) [87] | For polishing the working electrode surface to ensure a fresh, reproducible, and contaminant-free surface. |
| Test Resistor (10 kΩ) [87] | Used for the initial diagnostic check of the potentiostat and cables. |
| Quasi-Reference Electrode (e.g., bare silver wire) [87] | A simple alternative to a commercial reference electrode for troubleshooting reference electrode failures. |
A methodical approach to troubleshooting is indispensable for reliable cyclic voltammetry data. By first verifying the potentiostat and cables with a simple resistor test, then systematically evaluating each electrode, researchers can efficiently isolate and rectify the root cause of common experimental issues. Adhering to this protocol and maintaining properly prepared electrodes will minimize artifacts and ensure that the collected voltammograms accurately reflect the redox chemistry under investigation, thereby strengthening the foundation for subsequent data interpretation and scientific conclusions.
Diagnosing and Fixing Voltage and Current Compliance Errors
In the context of a broader thesis on cyclic voltammetry (CV) for redox reaction analysis, understanding and mitigating compliance errors is fundamental to obtaining reliable electrochemical data. Voltage and current compliance errors represent the operational limits of a potentiostat, the primary instrument used in CV experiments. Voltage compliance is reached when the instrument cannot maintain the desired potential between the working and reference electrodes, often due to high current flow or excessive solution resistance. Current compliance occurs when the measured current exceeds the instrument's maximum allowable range, potentially leading to signal clipping and loss of electrochemical information [11]. These errors are particularly critical in drug development, where precise characterization of redox-active pharmaceutical compounds—such as their metabolic oxidation pathways or reactive oxygen species generation—depends on highly accurate voltammetric measurements. Even minor compliance limitations can distort key parameters including peak potentials, peak currents, and voltammetric shape, leading to incorrect interpretation of electron transfer kinetics and reaction mechanisms [41].
Theoretical Background: The Origin of Compliance Limits
Electrochemical compliance limits are intrinsically linked to the fundamental equation governing potentiostat operation. The instrument must apply a potential (Eapplied) sufficient to overcome both the desired interfacial potential (Einterface) and the ohmic drop (iRu) across the solution: Eapplied = Einterface + iRu, where i is the current and R_u is the uncompensated solution resistance [11]. As either the current or the solution resistance increases, the required output voltage rises accordingly. When this demand exceeds the potentiostat's voltage compliance specification, control of the working electrode potential is lost, and the resulting voltammogram becomes distorted.
Similarly, the current compliance limit is encountered when the faradaic process generates more electrons than the instrument can accurately measure. The peak current in a reversible cyclic voltammogram is given by the Randles-Ševčík equation: i_p = (2.69 × 10^5) n^(3/2) A C D^(1/2) ν^(1/2) [11] where n is the number of electrons, A is the electrode area (cm²), C is the concentration (mol/cm³), D is the diffusion coefficient (cm²/s), and ν is the scan rate (V/s). From this relationship, it is evident that high concentrations of redox-active species, large electrode surfaces, or fast scan rates can generate currents that exceed the instrument's measurable range, particularly in applications involving highly conductive drug molecules or catalytic materials.
Diagnosing Compliance Errors
Visual Signatures in Voltammograms
Recognizing the characteristic shapes of compliance errors in cyclic voltammograms is the first step in diagnosis.
Voltage Compliance Error Signature: Appears as a flattening or clipping of the current response at the vertex potentials, where the scan direction reverses. The system cannot maintain the required potential, causing the current to saturate. This distortion is often accompanied by an abnormally large peak separation (ΔE_p >> 59/n mV) that increases with scan rate, mimicking slow electron transfer kinetics but originating from instrumental limitation rather than chemical behavior [11].
Current Compliance Error Signature: Manifests as a truncation of the current peaks throughout the voltammogram, where the current response reaches a plateau at the instrument's maximum measurable value rather than displaying the characteristic symmetric duck shape. This is particularly prevalent when studying high-concentration solutions or highly catalytic systems common in pharmaceutical screening [11].
Quantitative Diagnostic Parameters
Systematic analysis of voltammetric parameters can help distinguish true electrochemical behavior from artifact.
Table 1: Diagnostic Parameters for Compliance Errors
| Parameter | Reversible System (Benchmark) | Voltage Compliance Error | Current Compliance Error |
|---|---|---|---|
| ΔE_p (peak separation) | ≈59/n mV [11] | Significantly >59/n mV, increases disproportionately with scan rate | Approximately 59/n mV, but peaks truncated |
| ipa/ipc (peak current ratio) | ≈1 [11] | Often <1, decreases with increasing scan rate | <1 if clipping affects one peak more than the other |
| i_p vs. ν^(1/2) plot | Linear [11] | Non-linear, deviates at higher scan rates | Linear until compliance limit, then plateaus |
| Background charging current | Proportional to scan rate (ic = νCdl) [11] | Excessive charging current contributes to voltage compliance issues | Unaffected unless voltage compliance is also triggered |
Experimental Protocols for Error Mitigation
Protocol 1: Optimization of Electrochemical Cell Configuration
Objective: Minimize uncompensated resistance (R_u) to prevent voltage compliance errors.
Electrode Positioning: Place the Luggin capillary tip correctly at a distance of approximately 2 times its diameter from the working electrode surface. This configuration minimizes ohmic drop without significantly shielding the working electrode. Materials: Luggin capillary, three-electrode cell (working, reference, counter) [11].
Electrolyte Selection: Use a supporting electrolyte at sufficient concentration (typically ≥0.1 M) to ensure high ionic conductivity. For non-aqueous systems in drug development (e.g., acetonitrile or DMF), use tetrabutylammonium hexafluorophosphate (NBu₄PF₆) at 0.1 M concentration [89]. Rationale: High electrolyte concentration reduces solution resistance, thereby decreasing iR_u drop and the voltage demand on the potentiostat.
Electrode Surface Area: For highly concentrated redox-active solutions, consider using a microelectrode (diameter <50 μm) instead of conventional macroelectrodes. The small area significantly reduces total current (according to the Randles-Ševčík equation) while maintaining current density, thus preventing current compliance issues [11].
Protocol 2: Instrument Parameter Calibration and Validation
Objective: Configure potentiostat settings to operate within compliance boundaries while maintaining signal quality.
Current Range Selection:
- Begin with the lowest current range compatible with expected signals.
- Run a blank voltammogram with only electrolyte solution to measure background current.
- Gradually increase analyte concentration or scan rate while monitoring for clipping.
- If clipping occurs, switch to a higher current range before signal acquisition.
Positive Feedback iR Compensation:
- Many modern potentiostats offer electronic iR compensation.
- First, measure R_u using current-interrupt or electrochemical impedance methods.
- Apply compensation gradually (start with 70-80% of measured R_u) to avoid circuit oscillation.
- Critical Note: Over-compensation can lead to instrument instability and distorted voltammograms [11].
Scan Rate Optimization:
- Perform preliminary experiments across a range of scan rates (e.g., 10-1000 mV/s).
- Plot i_p versus ν^(1/2) to identify where linearity is lost, indicating compliance limitations.
- Select scan rates that fall within the linear region for quantitative kinetic analysis [11].
Protocol 3: Systematic Diagnosis Workflow
The following diagnostic pathway provides a logical approach to identifying and resolving compliance errors:
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for Compliance-Free Cyclic Voltammetry
| Item | Function/Benefit | Application Notes |
|---|---|---|
| Tetrabutylammonium Hexafluorophosphate (NBu₄PF₆) | High-concentration (≥0.1 M) supporting electrolyte reduces solution resistance (R_u), minimizing voltage compliance errors [89]. | Preferred for non-aqueous studies (acetonitrile, DMF) of drug molecules; wide electrochemical window. |
| Potassium Chloride (KCl) | High-concentration (≥0.1 M) aqueous supporting electrolyte with excellent conductivity for aqueous biological systems [11]. | Suitable for water-soluble compounds; use caution with silver reference electrodes (AgCl formation). |
| Luggin Capillary | Positions reference electrode proximity to working electrode without current shielding, minimizing uncompensated resistance [11]. | Critical for accurate potential control; maintain distance of ~2x capillary diameter from working electrode. |
| Microelectrodes (e.g., Pt, Au, Glassy Carbon) | Small surface area (<50 μm diameter) reduces absolute current, preventing current compliance issues while maintaining current density [11]. | Ideal for high concentration samples or fast scan rates; enables work in low electrolyte conditions. |
| Ferrocene/Ferrocenium Redox Couple | Internal potential standard for non-aqueous systems; validation tool for compliance-free operation [89]. | After modifications, test system with 1 mM ferrocene; reversible voltammogram (ΔE_p ≈ 59 mV) confirms compliance resolution. |
| Potassium Ferricyanide | Aqueous redox standard for validating system performance and confirming absence of compliance errors [11]. | Use at 1 mM concentration in 1 M KCl; well-characterized reversible behavior serves as benchmark. |
Advanced Applications: DFT Calculations and Mechanistic Analysis
For researchers incorporating computational methods, compliance errors present particular challenges as they introduce discrepancies between theoretical predictions and experimental data. Recent studies bridge this gap by calibrating density functional theory (DFT) calculations with high-quality experimental voltammetry [41]. The scheme of squares framework provides a systematic approach to diagram possible electron transfer (ET) and proton transfer (PT) pathways, differentiating between decoupled ET-PT and concerted proton-electron transfer (PET) mechanisms [41]. When compliance errors distort experimental voltammograms, this calibration becomes unreliable, potentially misassigning reaction pathways of pharmaceutical compounds. For instance, accurately determining the redox potential of drug candidates using the Nernst equation (E = E⁰ + (RT/nF)ln([Ox]/[Red])) requires compliance-free data to ensure proper correlation with computed Gibbs free energy changes (ΔG = -nFE⁰) [41].
Voltage and current compliance errors represent significant but manageable challenges in cyclic voltammetry research, particularly in drug development where precise electrochemical characterization is essential. Through systematic diagnosis using the visual and quantitative parameters outlined in this work, followed by implementation of the optimized experimental protocols, researchers can effectively mitigate these instrumental limitations. The integration of proper cell configuration, judicious instrument settings, and validation with standard redox couples ensures the acquisition of high-quality voltammetric data. This rigorous approach to compliance management forms the foundation for reliable correlation between experimental results and computational models, ultimately advancing the understanding of redox mechanisms in pharmaceutical and biological systems.
Cyclic Voltammetry (CV) is a powerful and versatile electrochemical technique used to study the behavior, kinetics, and mechanisms of electrochemical reactions at the electrode/electrolyte interface [90]. By applying a linearly varying potential to an electrochemical cell and measuring the resulting current, researchers can obtain rich information about redox processes, which is indispensable in fields like materials science, chemistry, and drug development [90]. A typical cyclic voltammogram presents a characteristic "duck-shaped" plot for a reversible system, with key parameters including anodic peak current (ipa), cathodic peak current (ipc), anodic peak potential (Epa), and cathodic peak potential (Epc) [11] [9].
However, even with modern potentiostats, experimental results can be marred by anomalies that distort the voltammogram and complicate data interpretation. Common issues include excessive noise, sloping baselines, and hysteresis, often stemming from relatively small mistakes in experimental setup or fundamental electrochemical processes [87]. This application note, framed within a broader thesis on CV for redox reaction analysis, provides detailed protocols for diagnosing, troubleshooting, and resolving these specific challenges to ensure data integrity and reliability.
Understanding and Mitigating Noise
In electrochemical measurements, noise is defined as any unwanted disturbance that obscures the desired faradaic signal. It can be categorized as either random noise or systematic noise [91]. Random noise, as demonstrated in studies using carbon-fiber electrodes, can originate from the potentiostat's internal electronics, such as Johnson noise from the feedback resistor of the current transducer [92]. Systematic noise often includes line noise (50/60 Hz pickup from mains electricity) and noise arising from the physiological activity of a test subject in in vivo studies [92].
Experimental Protocols for Noise Reduction
Protocol 2.2.1: Comprehensive Noise Minimization
- Shielding and Cabling: Use shielded cables for all electrode connections. In demonstrations, unshielded cables act as antennas, picking up significant environmental noise. A Faraday cage can be highly effective at eliminating this external interference [91].
- Mains Frequency Synchronization: Configure the potentiostat to start each cyclic voltammogram either in phase or 180 degrees out of phase with the line frequency. This technique helps discriminate against line noise [92].
- Filtering and Averaging: Employ appropriate analog and digital filtering. Improve the signal-to-noise ratio by using ensemble averaging of multiple repetitive cyclic voltammograms [92].
- Parameter Optimization: Adjust the step potential (or sampling threshold). A very large step potential can make the voltammogram appear sharp and jagged, while a smaller step potential (e.g., ≤ 5 mV) produces smoother curves, though it generates larger data files [93].
- Connection Integrity: Ensure all electrodes are properly connected and submerged. A poor connection to the working electrode can result in a very small, noisy current, as only residual current from the potentiostat circuitry is detected [87].
Table 1: Strategies for Noise Mitigation and Their Applications
| Strategy | Protocol | Primary Noise Type Addressed | Key Consideration |
|---|---|---|---|
| Shielding | Use shielded cables; Enclose setup in a Faraday cage [91] | Systematic (Line noise, environmental pickup) | Essential for low-current or high-impedance measurements. |
| Synchronization | Start CV scan in/out of phase with mains frequency [92] | Systematic (Line noise) | Requires potentiostat software capable of this timing. |
| Averaging | Acquire and ensemble average multiple CV cycles [92] | Random | Increases total experiment time; assumes system is stable. |
| Digital Filtering | Apply post-measurement smoothing in software [91] | Random & Systematic | Can distort kinetic information if over-applied. |
| Parameter Adjustment | Use a smaller sampling threshold/step potential [93] | Quantization/Algorithmic | Balance between data smoothness and file size. |
Figure 1: Diagnostic workflow for identifying and resolving sources of noise in cyclic voltammetry.
Diagnosing and Correcting Sloping Baselines
Origins of Non-Ideal Baselines
A non-flat, or sloping, baseline deviates from the ideal horizontal line in the double-layer region and can severely impact the accurate calculation of charge and peak parameters. For instance, when determining the electrochemical surface area (ECSA) of platinum on carbon (Pt/C) catalysts, a skewed baseline in the double-layer region significantly affects the charge calculation in the hydrogen adsorption/desorption regions [94]. Unlike pure platinum, which has a horizontal baseline, the Pt/C catalysts exhibit a skewed baseline due to surface functional groups from the carbon support [94]. This skewness is often not identical for the cathodic and anodic scans, leading to different calculated charges for adsorption and desorption. Furthermore, the baseline can change during accelerated stress tests (AST) due to oxidation (corrosion) of the carbon support, potentially leading to a ten-fold greater error in the H-desorption area compared to the H-adsorption area [94].
More generally, problems with the working electrode itself can lead to a non-straight baseline, although additional capacitive processes at the electrodes with currently unknown origins can also be a cause [87].
Protocol for Baseline Analysis and Correction
Protocol 3.2.1: Handling Sloping Baselines in Pt/C ECSA Determination
- Background Acquisition: Always run a background CV in the pure electrolyte (without analyte) using identical parameters (electrode, potential window, scan rate).
- Baseline Selection: For Pt/C catalysts, avoid using a simple horizontal baseline. Instead, use the current response from the corresponding carbon support material (without the metal catalyst) as a closer approximation of the true baseline [94].
- Charge Calculation: Use the charge associated with the hydrogen adsorption (H~ad~) region, rather than the desorption (H~de~) region, for ECSA calculation. The H~de~ charge is more significantly affected by changes in the carbon support during durability studies [94].
- Monitor Changes: During durability studies (ASTs), be aware that the baseline contribution will likely increase with the number of stress cycles due to carbon corrosion. Using a constant baseline from the initial cycle can lead to substantial errors in ECSA loss calculations [94].
Table 2: Common Causes and Solutions for Sloping Baselines
| Cause of Sloping Baseline | Effect on Voltammogram | Recommended Correction Protocol |
|---|---|---|
| Carbon Support Functional Groups [94] | Skewed, non-horizontal double-layer region. | Use baseline from carbon support; Prefer H~ad~ charge for ECSA. |
| Carbon Corrosion during AST [94] | Increasing baseline slope over successive cycles. | Account for evolving baseline; do not use a constant baseline. |
| Working Electrodefaults [87] | Non-straight, unstable baseline. | Polish and clean the working electrode (see Protocol 4.2.1). |
| Unknown Capacitive Processes [87] | Unpredictable baseline shape. | Acquire background scan and subtract from sample voltammogram. |
Resolving Hysteresis in the Baseline
Understanding Hysteresis and Charging Currents
Hysteresis in the baseline, where the forward and reverse scans do not overlap and form a "loop" even in regions without faradaic activity, is primarily due to charging currents at the electrode-solution interface [87]. This interface behaves like a capacitor, which must be charged before an electrochemical process can occur. The magnitude of this charging current is directly proportional to the scan rate (v) and the double-layer capacitance (C~dl~) [11]. Additional, often larger, charging currents can be caused by faults in the working electrode, such as poor internal contacts or poor seals, which can lead to high resistivity and capacitance, manifesting as a pronounced hysteresis and a sloping baseline [87].
Experimental Strategies to Minimize Hysteresis
Protocol 4.2.1: General Troubleshooting and Electrode Maintenance The following procedure, inspired by general troubleshooting guidelines, helps identify whether the issue lies with the potentiostat, cables, or electrodes [87].
- Potentiostat and Cable Check:
- Disconnect the electrochemical cell.
- Connect a 10 kΩ resistor between the working electrode input and the combined reference and counter electrode inputs.
- Run a CV scan (e.g., from +0.5 V to -0.5 V). The result should be a straight line obeying Ohm's law (V=IR). Any deviation indicates a problem with the potentiostat or cables [87].
- Reference Electrode Check:
- Set up the electrochemical cell as normal, but connect the reference electrode cable to the counter electrode (so the RE and CE are shorted).
- Run a linear sweep voltammetry experiment with your analyte. A standard, though potential-shifted and slightly distorted, voltammogram should be obtained. If not, the issue is likely with the working or counter electrodes. If a standard voltammogram is obtained, the problem lies with the reference electrode (e.g., a blocked frit) [87].
- Working Electrode Care:
- Polishing: Mechanically polish the working electrode (e.g., glassy carbon) with progressively finer alumina slurries (down to 0.05 μm) and wash it thoroughly to remove adsorbed species [87].
- Electrochemical Cleaning: For a Pt electrode, clean it by cycling the potential in a 1 M H~2~SO~4~ solution between the potentials for H~2~ and O~2~ evolution [87].
- Parameter Adjustment: To reduce the intrinsic charging current, decrease the scan rate, increase the concentration of the analyte, or use a working electrode with a smaller surface area [87].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials and Reagents for Reliable Cyclic Voltammetry
| Item | Function / Purpose | Example & Notes |
|---|---|---|
| Three-Electrode System | Fundamental setup for controlled potential application [90]. | Working (e.g., Glassy Carbon), Reference (e.g., Ag/AgCl), Counter (e.g., Pt wire). |
| Potentiostat | Applies potential and measures current [87]. | Instruments from Ossila, Pine Research, etc. Modern versions have 16-bit DAC for less distortion [93]. |
| Supporting Electrolyte | Carries current and minimizes migration and ohmic drop [87]. | 0.1 M KCl, TBAPF~6~ in organic solvents. Must be inert and highly purified. |
| Internal Standard | Validation and potential calibration of the setup [9]. | Ferrocene (Fc/Fc⁺ couple) in non-aqueous electrolytes. |
| Electrode Polishing Kit | Ensures reproducible, clean electrode surface [87]. | Alumina or diamond slurries (1.0, 0.3, and 0.05 μm) on a microcloth pad. |
| Electrochemical Cell | Container for the analyte solution and electrodes [87]. | Can be a dedicated glass cell or a vial; must allow proper electrode placement. |
| Faraday Cage | Shields the setup from external electromagnetic noise [91]. | A grounded metal enclosure. Essential for low-detection limit measurements. |
| Test Resistor/Chip | Verifies proper function of potentiostat and cables [87]. | A 10 kΩ resistor or a manufacturer-supplied test cell chip (e.g., Ossila). |
Unusual voltammograms featuring noise, sloping baselines, and hysteresis are common challenges in electrochemical research. By applying the systematic diagnostic and mitigation strategies outlined in this note—ranging from proper shielding and grounding to careful baseline selection and electrode maintenance—researchers can significantly improve the quality of their CV data. Adherence to detailed experimental protocols is crucial for obtaining reliable and reproducible results, which form the foundation for accurate analysis of redox reactions in fields ranging from drug development to materials science. A meticulous approach to troubleshooting not only resolves immediate issues but also deepens the practitioner's understanding of the underlying electrochemical principles.
Identifying and Eliminating Unexpected Peaks from Impurities and System Artifacts
In cyclic voltammetry (CV), the appearance of unexpected peaks can significantly complicate the interpretation of electrochemical data. These anomalous signals often stem from electrochemical impurities or system artifacts, potentially leading to incorrect conclusions about the redox properties of the system under study. This application note provides a structured framework for identifying the source of these peaks and implementing protocols for their elimination, with a specific focus on trace metal contamination and instrumental artifacts. The guidance is particularly crucial for researchers in pharmaceutical development where electrochemical characterization informs drug stability, reactivity, and metabolic profiling.
A fundamental understanding of expected CV behavior provides the baseline for identifying anomalies. For a reversible redox couple, the voltammogram exhibits symmetric anodic and cathodic peak currents with a separation (ΔEp) of approximately 59/n mV at 25°C, and peak potentials that remain independent of the scan rate [95]. Deviations from this ideal behavior, such as large peak separations, shifted potentials, or the presence of additional, unaccounted-for peaks, often indicate the influence of impurities, slow electron transfer kinetics, or coupled chemical reactions [95].
Systematic Approach to Peak Identification
A systematic investigation is required to diagnose the origin of unexpected voltammetric peaks. The process involves correlating the observed electrochemical response with potential chemical and physical sources.
Investigator's Checklist for Unexpected Peaks
- Correlate Peaks with Sample Composition: Review the chemical composition of the electrolyte, including the solvent, supporting electrolyte, and analyte. Identify any chemical species that could produce a redox signal in the applied potential window.
- Interrogate the Electrode Surface: Examine the working electrode for surface contamination, adsorption of species, or changes in morphology. A contaminated electrode can introduce faradaic processes unrelated to the bulk analyte.
- Analyze Scan Rate Dependence: Perform CV measurements at multiple scan rates. Peaks originating from diffusing redox species will show a proportional relationship between peak current and the square root of the scan rate. Peaks from surface-bound species will show a linear relationship between peak current and scan rate [96].
- Benchmark Against a Control: Run a CV of the background electrolyte (lacking the analyte) using the same experimental setup. Any peaks appearing in this control measurement are unequivocally linked to the electrolyte or electrode system itself.
- Evaluate Reproducibility: Assess the reproducibility of the unexpected peak across multiple experimental replicates and different sample preparations. Non-reproducible peaks may indicate sporadic contamination.
Common Culprits and Their Signatures
Unexpected peaks generally arise from two primary categories: chemical impurities and system artifacts. The table below summarizes common sources and their characteristic electrochemical signatures.
Table 1: Common Sources of Unexpected Peaks in Cyclic Voltammetry
| Source Category | Specific Source | Characteristic Voltammetric Signature |
|---|---|---|
| Chemical Impurities | Trace Metals (e.g., Fe in alkaline electrolytes) | Enhanced OER activity, growth of specific metal hydroxide phases (e.g., γ-NiOOH), and appearance of new redox peaks after continuous cycling [97]. |
| Oxygen Contamination | Irreversible reduction peak near the cathodic solvent limit; disappears after purging with an inert gas [95]. | |
| Organic Contaminants | Often manifests as broad, irreversible peaks; can foul the electrode surface, reducing electron transfer kinetics. | |
| System Artifacts | Uncompensated Resistance (Ru) | Peak broadening, increased ΔEp, and shifting of peak potentials with increasing current [97]. |
| Reference Electrode Issues | Drifting potentials and inconsistent peak positions between experiments. | |
| Instrumental Ground Loops | 60/50 Hz noise superimposed on the voltammogram, producing a "zig-zag" pattern. |
Experimental Protocols
Protocol 1: Detection of Trace Iron Impurities in Alkaline Electrolytes
This protocol is adapted from a established method for detecting Fe impurities using a nickel working electrode, which is highly susceptible to incorporating Fe into its oxide matrix [97].
1. Principle: Trace Fe impurities in KOH or NaOH electrolytes incorporate into the growing Ni(OH)₂/NiOOH film during potential cycling. This incorporation alters the phase behavior of the nickel oxide and enhances the current density for the oxygen evolution reaction (OER), providing a clear diagnostic for contamination.
2. Materials and Reagents: Table 2: Research Reagent Solutions for Iron Impurity Detection
| Item | Function | Specification |
|---|---|---|
| Nickel Foam Electrode | Working electrode | ~1 cm² geometric area, with Ti current collector [97]. |
| PTFE Electrochemical Cell | Houses the electrolyte | Prevents contamination from glass leaching [97]. |
| Graphite Rod | Counter electrode | Inert electrode for alkaline environments [97]. |
| Hg/HgO Reference | Reference electrode | Standard for alkaline electrolytes [97]. |
| KOH or NaOH Electrolyte | Electrolyte under test | 1 M, prepared with high-purity water [97]. |
| PDMS (Sylgard 184) | Electrode coating | Insulates parts of the Ni foam not intended for exposure [97]. |
3. Procedure: 1. Electrode Preparation: Coat a Ni foam electrode (1 cm²) with PDMS, leaving only the desired surface area exposed. Cure the PDMS according to the manufacturer's instructions [97]. 2. Cell Assembly: Assemble the three-electrode system in a PTFE cell filled with the test electrolyte. 3. Potentiostat Sequence: * Open Circuit Potential (OCP): Measure for 5-10 minutes to stabilize the system [97]. * iR Compensation: Measure the uncompensated resistance (Ru) via potentiostatic EIS and apply 85% positive feedback iR compensation for all subsequent steps [97]. * Initial Assessment: Record 3 repeated LSV scans and 2 CV scans from 0.15 to 0.8 V vs. Hg/HgO at 50 mV/s [97]. * CV Conditioning (Activation): Run 2000 cycles of CV using the same parameters as the initial assessment [97]. * Intermediate Scans: Every 100 cycles, record a slow-scan CV (e.g., 5 mV/s) to monitor spectral evolution clearly [97]. * Final Assessment: Repeat the LSV and CV scans from the initial assessment [97].
4. Data Interpretation:
- Fe-Free Electrolyte: The CV will show distinct, stable redox peaks for the β-NiOOH phase and two anodic peaks at approximately +1.54 V~RHE~ and +1.65 V~RHE~. The OER current density at high potentials remains relatively low [97].
- Fe-Contaminated Electrolyte: The CV will show a significant increase in OER current density, a shift in the γ-phase peak, and the disappearance of the anodic peaks at +1.54 V~RHE~ and +1.65 V~RHE~ [97].
The workflow for this protocol is outlined below.
Protocol 2: Mitigation of System Artifacts
This protocol addresses common instrumental and setup-related artifacts that can introduce spurious features in CV data.
1. Minimizing Uncompensated Resistance (Ru): * Positioning: Place the Luggin capillary correctly to minimize the distance between the reference and working electrodes without shielding the electrode surface. * iR Compensation: Always use the positive feedback iR compensation mode when possible, especially for systems with high current or low electrolyte conductivity [97]. The resistance value (Ru) should be determined experimentally via electrochemical impedance spectroscopy (EIS) prior to CV measurements. * Supporting Electrolyte: Use a sufficient concentration of supporting electrolyte (typically 0.1 - 0.5 M) to ensure ionic conductivity.
2. Ensuring Electrolyte and System Purity: * Oxygen Removal: Sparge the electrolyte with high-purity nitrogen or argon for at least 15-20 minutes prior to measurements. Maintain a positive pressure of inert gas over the solution during experiments. * Electrode Cleaning: Polish the working electrode meticulously with alumina slurry (e.g., 0.3 μm and 0.05 μm) on a microcloth pad, followed by sonication and rinsing with purified solvent [96]. * High-Purity Reagents: Use the highest grade available for solvents, salts, and analytes.
Data Analysis and Decision Workflow
When an unexpected peak is observed, follow the logical pathway below to diagnose its origin.
The reliable interpretation of cyclic voltammetry data in redox reaction analysis hinges on the analyst's ability to distinguish between signals from the target analyte and those arising from impurities and artifacts. By employing the systematic identification checklist, implementing the specific protocols for impurity detection, and rigorously applying mitigation strategies for system artifacts, researchers can significantly enhance the quality and reproducibility of their electrochemical data. This is particularly critical in fields like drug development, where accurate electrochemical characterization can directly impact the understanding of a compound's stability and reactivity. Adopting a standardized, FAIR-compliant workflow for data generation and management, as highlighted in recent open-source approaches, further ensures the integrity and reusability of electrochemical data [98].
Within the framework of cyclic voltammetry for redox reaction analysis, the integrity of the working electrode surface is paramount. The sensitivity, reproducibility, and overall validity of electrochemical data are contingent upon a pristine and well-defined electrode surface. Contaminants or microscopic imperfections can significantly alter electron transfer kinetics, leading to distorted voltammograms, shifted peak potentials, and unreliable quantification of redox-active species, such as pharmaceutical compounds in drug development research [53]. This application note provides detailed, actionable protocols for the maintenance, polishing, and cleaning of working electrodes to ensure reproducible and high-fidelity results in electrochemical analysis.
The Scientist's Toolkit: Essential Materials for Electrode Polishing
The following table catalogues the essential reagents and materials required for effective electrode maintenance, as derived from established polishing procedures [99].
Table 1: Key Research Reagent Solutions and Materials for Electrode Polishing
| Item Name | Function and Purpose |
|---|---|
| Emery Paper (e.g., UF800) | Used for initial rough polishing to remove large depressions and imperfections from the electrode surface. |
| Polishing Diamond (Suspension) | An abrasive for intermediate polishing, creating a smooth, pre-finished surface on hard materials like glassy carbon. |
| Polishing Alumina (Suspension) | A fine abrasive used for the final polishing step to achieve a mirror-finish, essential for reproducible electron transfer. |
| Diamond Polishing Pad | A dedicated pad used in conjunction with diamond suspension for the intermediate polishing step. |
| Alumina Polishing Pad (e.g., brown felt pad) | A dedicated soft pad used with alumina suspension for the final, high-gloss finish. |
| Distilled Water | Used for rinsing the electrode and wetting polishing pads between steps to prevent cross-contamination of abrasives. |
| Acetone | A solvent used to wipe the electrode surface to remove residual polishing abrasives and organic contaminants. |
| Nitric Acid (e.g., 6N) | A specific reagent for the removal of old mercury amalgam from gold electrodes before re-polishing. |
| Soft Tissue/ Lint-free Wipes | For gently drying and wiping the electrode surface after polishing and cleaning without introducing scratches. |
Experimental Protocols for Electrode Polishing and Regeneration
This section provides step-by-step methodologies for maintaining different types of working electrodes commonly used in cyclic voltammetry.
General Polishing Procedure for Glassy Carbon Electrodes
Glassy carbon is a widely used electrode material due to its broad potential window and inertness. Its performance is highly dependent on surface finish [99]. The polishing process involves three distinct stages, though not all are always necessary.
Table 2: Polishing Steps and Parameters for Glassy Carbon Electrodes
| Step | Purpose | Abrasive & Pad | Key Parameters and Instructions |
|---|---|---|---|
| 1. Rough Polishing | Remove large surface depressions and defects. | Emery Paper (e.g., UF800) with distilled water. | - Polish on a hard, flat surface.- Hold the electrode perpendicular to the paper.- Apply light force and move in a figure-8 motion for 30 seconds to 2 minutes.- Rinse thoroughly with distilled water. |
| 2. Intermediate Polishing | Create a smooth, pre-finished surface. | Polishing Diamond (8-10 drops) on a diamond polishing pad. | - Shake the diamond suspension well.- Polish in a circular motion for approximately 2 minutes until the surface shines.- Rinse the electrode and wipe with acetone to remove all diamond abrasive. |
| 3. Finish Polishing | Achieve a mirror-like finish for optimal electron transfer. | Polishing Alumina (5-6 drops) on an alumina polishing pad wetted with 10-20 drops of water. | - Polish for 3 to 4 minutes.- Rinse the electrode thoroughly with distilled water to remove all alumina particles.- Wipe the surface gently with a soft, lint-free tissue and air dry. |
Protocol for Gold Amalgam Electrode Regeneration
Thin-layer mercury amalgam electrodes are valuable for detecting reducible species, such as thiols, but require specific maintenance [99].
Removal of Old Amalgam:
- Apply 6N nitric acid drops to the electrode surface. The amalgam will dissolve, changing color from gray-black to yellow.
- If the reaction is slow, replace the nitric acid. Gently scrape the surface with a glass pipette tip while applying the acid.
- Once the color changes, rinse extensively with distilled water.
Regeneration of Gold Electrode Surface:
- Follow the three-step polishing procedure outlined in Section 3.1 (Rough, Intermediate, and Finish Polishing) to achieve a mirror-like finish on the underlying gold electrode.
Preparation of Fresh Mercury Amalgam:
- Place a small amount of mercury onto the freshly polished gold surface, ensuring complete coverage.
- After 2-3 minutes, carefully scrape off the excess mercury.
- Wipe the surface gently with a soft, dry tissue until it shines like a mirror.
- Crucially, allow the newly formed amalgam electrode to stabilize for at least 6 hours before use. Using it immediately will result in a high background and unstable response [99].
Workflow for Electrode Maintenance
The following diagram illustrates the logical decision-making process and workflow for maintaining electrodes based on their type and condition.
Data Presentation: Polishing Parameter Optimization
The efficacy of the polishing process can be influenced by specific parameters. The table below summarizes key considerations for different EDC methods, which inform the manual polishing approach [100].
Table 3: Operational Parameters for Surface Modification Techniques
| Parameter | Ti-Powder Suspension | Ti- Electrode | 3DPE (Ti6Al4V) |
|---|---|---|---|
| Tool Electrode | Titanium Powder | Titanium | Ti6Al4V |
| Peak Current (Ia) | 8, 10, 12 A | 8, 10, 12 A | 6, 8, 10 A |
| Pulse On Time (Ton) | 120, 100, 80 μs | 120, 100, 80 μs | 120 μs |
| Pulse Off Time (Toff) | 60, 40, 20 μs | 60, 40, 20 μs | 60 μs |
| Machining Time | 15 min | 10 min | 10 min |
| Polarity | + (Positive) | - (Negative) | - (Negative) |
Adherence to the detailed polishing and cleaning protocols outlined in this document is a critical prerequisite for obtaining reliable and reproducible data in cyclic voltammetry studies of redox reactions. A consistently maintained electrode surface minimizes experimental artifacts, enabling accurate interrogation of reaction mechanisms and precise quantification of analytes, which is fundamental to advancing research in drug development and materials science.
In the analysis of redox reactions using cyclic voltammetry (CV), the faradaic current originating from electron transfer events is the primary signal of interest. However, this signal is invariably accompanied by non-faradaic, background currents, primarily from charging currents associated with double-layer capacitance and background currents from surface redox processes on the electrode itself [101] [102]. These parasitic currents can obscure the target faradaic signal, reducing the signal-to-noise ratio (SNR), limiting detection sensitivity, and complicating data interpretation. This Application Note details advanced, practical strategies—encompassing frequency-domain analysis, machine-learning-driven waveform design, and digital signal processing—to effectively separate and suppress these interfering currents, thereby enhancing the fidelity of electrochemical measurements.
Theoretical Background: The Nature of Interfering Currents
The Charging Current
The electrical double-layer at the electrode-solution interface behaves as a capacitor. When the applied potential changes during a voltammetric scan, current flows to charge or discharge this capacitor. This charging current ((ic)) is described by: (ic = C{dl} \times \frac{dE}{dt} ) where (C{dl}) is the double-layer capacitance and (\frac{dE}{dt}) is the scan rate [102]. The charging current is a fundamental, unavoidable consequence of potential sweep methods and scales linearly with scan rate.
The Background Current
Background currents often arise from persistent faradaic processes, such as the continuous oxidation and reduction of functional groups on the electrode surface or the presence of electroactive impurities in the electrolyte [103]. Unlike charging currents, their shape in a cyclic voltammogram often mirrors the underlying surface redox process and can be highly dependent on the electrode material and its history.
Table 1: Key Characteristics of Interfering Currents
| Current Type | Origin | Dependence on Scan Rate | Primary Influence on CV |
|---|---|---|---|
| Charging Current | Double-layer capacitance | Linear (( \propto \frac{dE}{dt} )) | Obscures redox peak shape, increases baseline slope |
| Background Current | Surface redox reactions | Complex (often non-linear) | Contributes to a sloping, non-uniform baseline |
Strategy 1: Frequency-Domain Analysis and Waveform Optimization
Traditional time-domain analysis often struggles to disentangle overlapping faradaic and non-faradaic processes. A powerful alternative is to reframe the measurement in the frequency domain [101].
Protocol: Frequency-Structured Framework for Voltammetric Analysis
This protocol outlines a method to analyze voltammetric signals by their harmonic components to guide waveform optimization [101].
- Signal Acquisition: Acquire voltammetric data using a standard technique (e.g., AC voltammetry) across a range of excitation frequencies.
- Spectral Decomposition: Perform a Fast Fourier Transform (FFT) on the acquired current signal to decompose it from the time domain into its constituent frequency components.
- Process Separation: Identify the harmonic signatures of faradaic and capacitive processes. The modified Randles equivalent circuit model can be used to interpret the frequency-domain response, where charge-transfer and diffusion processes (faradaic) and double-layer charging (capacitive) exhibit different frequency-dependent behaviors [101].
- Spectral Descriptor Calculation: Quantify the signal's harmonic structure using descriptors such as:
- Centroid Frequency: The average frequency, weighted by power.
- Bandwidth: The range of frequencies present.
- Low-Frequency Power Fraction: The proportion of signal power in the low-frequency region where faradaic processes often dominate.
- Waveform Tuning: Use these spectral descriptors to select or design a waveform that maximizes faradaic visibility. This typically involves enhancing components in frequency bands where the faradaic signal is strong relative to the capacitive background.
Data Interpretation
Frequency-domain analysis reveals that increasing the excitation frequency generally suppresses faradaic clarity, as the slower faradaic processes cannot keep pace [101]. Quantitative spectral descriptors provide a direct link between waveform geometry and measurement performance, coalescing parameters that are incompatible in the time domain into a unified framework for optimization.
Strategy 2: Machine-Learning-Guided Waveform Design
The design of optimal voltammetric waveforms is challenging due to intractably large combinatorial search spaces. Bayesian optimization provides a data-driven solution to this problem [103].
Protocol: SeroOpt Workflow for Waveform Optimization
This protocol describes a machine-learning (ML) workflow to design waveforms optimized for specific analytes, such as the neurotransmitter serotonin [103].
- Define Objective Metric: Select a quantitative performance metric for optimization (e.g., detection accuracy for a target analyte, signal-to-noise ratio, or selectivity over an interferent).
- Initialize with Training Data: Collect a small initial set of experimental training data by testing a limited number of starting waveforms (e.g., random designs or historic performers).
- Train Surrogate Model: Use Bayesian optimization to build a probabilistic surrogate model that approximates the complex, unknown relationship between waveform parameters and the objective metric.
- Select New Waveform: Allow the ML algorithm to query the surrogate model and propose a new waveform predicted to maximize the objective function.
- Experimental Validation: Test the proposed waveform experimentally and measure its true performance.
- Iterative Refinement: Add the new result to the training dataset and repeat steps 3-5 for several iterations. The model improves with each cycle, rapidly converging on an optimized waveform.
Data Interpretation
The SeroOpt workflow has been shown to outperform both random searches and human-guided ("guess-and-check") waveform designs [103]. Interpretation of the ML-derived waveforms often reveals that the optimized parameters reflect established electrochemical domain knowledge, such as the importance of specific potentials and their sequence, validating the logic of the "black box" optimizer.
Table 2: Comparison of Waveform Design Strategies
| Strategy | Key Methodology | Advantages | Limitations |
|---|---|---|---|
| Human-Guided (Guess-Check) | Heuristics & one-parameter-at-a-time | Intuitive, based on experience | Inefficient; cannot explore large parameter spaces [103] |
| Frequency-Domain Guided | FFT & spectral metrics [101] | Provides principled framework for tuning | Requires conceptual awareness of spectral artefacts |
| ML-Guided (SeroOpt) | Bayesian optimization with experimental data [103] | Efficiently navigates large search spaces; data-driven | Requires initial training data and computational resources |
Strategy 3: Fast Fourier Transform (FFT) for Signal Enhancement
The FFT can be used not only for analysis but also as an integral part of the measurement technique to enhance the signal-to-noise ratio directly [104].
Protocol: FFT Continuous Cyclic Voltammetry (FFTCCV)
This protocol is adapted for the highly sensitive detection of organophosphates using a biosensor, but the core principles are widely applicable [104].
- Biosensor Preparation:
- Prepare a carbon-paste electrode modified with multiwall carbon nanotubes (MWCNTs).
- Immobilize the enzyme acetylcholinesterase (AChE) onto the electrode surface using a composite silicate sol-gel film.
- Flow-Injection System Setup:
- Integrate the biosensor into a flow-injection analysis (FIA) system. The flowing solution provides highly controllable mass transport conditions, improving reproducibility.
- FFTCCV Measurement:
- Apply a continuous cyclic voltammetry potential waveform.
- Subject the resulting current signal to Fast Fourier Transform.
- Enhance the SNR by integrating the signal in the frequency domain, where noise can be more effectively filtered.
- Parameter Optimization:
- Critical parameters such as enzyme activity, MWCNT quantity, and sweep rate should be optimized, for example, using Response Surface Methodology (RSM). The optimized sweeping rate reported for paraoxon detection is 10 V s¯¹ [104].
Data Interpretation
The FFTCCV method leverages the fact that the signal and noise often have different frequency distributions. By transforming the signal, integrating the key harmonic components, and filtering out-of-band noise, a significant enhancement in the SNR is achieved. This method enabled a detection limit for paraoxon as low as 6.2 × 10¯¹³ M [104].
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 3: Key Reagents and Materials for Electrochemical Noise Optimization
| Item | Function/Application |
|---|---|
| Carbon Fiber Microelectrodes | Standard working electrode for neurochemical analysis (e.g., in RPV); small size minimizes charging currents and allows in vivo sensing [103]. |
| Multiwall Carbon Nanotubes (MWCNTs) | Nanomaterial used to modify electrode surfaces; enhances electron transfer, provides high surface area, and serves as a carrier for enzyme immobilization [104]. |
| Acetylcholinesterase (AChE) | Enzyme used in inhibition-based biosensors for organophosphate pesticides; biological recognition element [104]. |
| Silicate Sol-Gel | Matrix for robust immobilization of enzymes (e.g., AChE) onto electrode surfaces, retaining bioactivity [104]. |
| Potentiostat (3-Electrode) | Instrument for applying potential waveforms and measuring resulting current; essential for all voltammetric techniques. |
| Bayesian Optimization Software | Machine learning code (e.g., SeroOpt) for navigating waveform parameter spaces and automating experimental optimization [103]. |
Effectively managing charging and background currents is paramount for advancing the sensitivity and reliability of cyclic voltammetry in redox reaction analysis. The strategies detailed herein—frequency-domain analysis, machine-learning-guided design, and FFT-based signal processing—provide a modern toolkit that moves beyond traditional empirical methods. By reconceptualizing the electrochemical signal from a temporal to a spectral construct, researchers can systematically design experiments that inherently maximize faradaic information while suppressing interference, ultimately leading to more precise and robust analytical data in fields ranging from neuroscience to environmental monitoring.
Validating Redox Mechanisms and Comparative Electrochemical Analysis
Cyclic voltammetry (CV) is a powerful electrochemical technique for probing redox reactions, providing critical information on formal potentials, electron-transfer kinetics, and reaction mechanisms. However, to fully characterize antioxidant properties, CV data must be correlated with complementary techniques that provide different perspectives on redox activity. The DPPH• (2,2-diphenyl-1-picrylhydrazyl) radical scavenging assay, monitored via spectrophotometry, serves as a vital complement to CV in antioxidant research. This combination provides a more comprehensive understanding of redox behavior by linking electrochemical oxidation potentials with practical radical quenching capabilities [105] [106].
This application note details the methodology for integrating these techniques, with specific focus on protocols for drug development researchers seeking to establish structure-activity relationships for novel antioxidant compounds. The correlation between these methods enables more accurate prediction of in vivo antioxidant behavior by connecting thermodynamic electrochemical parameters with kinetically-controlled radical scavenging efficiency [107].
Theoretical Background and Correlation Principles
Fundamental Mechanisms
Antioxidants neutralize free radicals through distinct mechanisms that can be probed by different analytical techniques:
- Hydrogen Atom Transfer (HAT): Involves concerted transfer of hydrogen atom to the radical
- Single Electron Transfer-Proton Transfer (SET-PT): Sequential electron transfer followed by proton transfer
- Sequential Proton-Loss Electron Transfer (SPLET): Deprotonation followed by electron transfer [106]
CV primarily detects SET-PT processes through oxidation potentials, while DPPH scavenging may proceed through any of these pathways depending on molecular structure and solvent conditions. The formal oxidation potential (E°) obtained from CV correlates with the compound's thermodynamic tendency to donate electrons, which directly influences its efficiency in DPPH radical reduction [106].
Correlation Between CV and DPPH Parameters
The relationship between electrochemical parameters and DPPH scavenging activity can be quantified through several key correlations:
- Oxidation Potential vs. IC₅₀: Compounds with lower oxidation potentials typically demonstrate lower IC₅₀ values in DPPH assays, indicating higher antioxidant potency
- Number of Redox Centers vs. TEAC: Molecules with multiple phenolic hydroxyl groups often show multiple oxidation waves in CV and higher Trolox Equivalent Antioxidant Capacity (TEAC)
- Reversibility of Oxidation vs. Antioxidant Stability: Irreversible oxidation in CV often correlates with decreased antioxidant stability in DPPH kinetic studies [106]
Figure 1: Relationship between analytical techniques and correlative parameters for antioxidant characterization
Experimental Protocols
Cyclic Voltammetry for Antioxidant Characterization
Principle: CV measures current response as the working electrode potential is swept linearly versus time, revealing redox potentials and electron transfer kinetics of antioxidant compounds [107].
Materials and Equipment:
- Potentiostat with three-electrode configuration
- Working electrode: Glassy carbon (3 mm diameter)
- Counter electrode: Platinum wire
- Reference electrode: Ag/AgCl (3 M KCl)
- Nitrogen gas for deaeration
- Supporting electrolyte: 0.1 M phosphate buffer (pH 7.4) or lithium perchlorate in organic solvents
Procedure:
- Prepare 1-5 mM antioxidant solution in appropriate solvent with supporting electrolyte
- Purge electrochemical cell with nitrogen for 10 minutes to remove oxygen
- Assemble three-electrode system with polished working electrode
- Set scan parameters: Initial potential = 0 V, switching potential = +1.0 V, final potential = 0 V
- Run multiple scan rates (20-500 mV/s) to assess diffusion control and reversibility
- Record voltammograms and determine formal potential (E°) as midpoint between anodic and cathodic peaks
Data Interpretation:
- Reversible systems: Peak separation ≈ 59/n mV, ipc/ipa ≈ 1
- Quasi-reversible: Peak separation > 59/n mV, scan-rate dependent
- Irreversible: No reverse peak, significant overpotential required
DPPH Radical Scavenging Assay with Spectrophotometric Detection
Principle: The stable DPPH radical exhibits deep purple color (λmax = 517 nm) which decays to yellow upon reduction by antioxidants, enabling spectrophotometric quantification of scavenging activity [105] [108].
Materials and Reagents:
- DPPH (2,2-diphenyl-1-picrylhydrazyl): Stable free radical source [108]
- Methanol or ethanol: Spectrophotometric grade solvent [109] [108]
- Trolox (6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid): Reference standard antioxidant [106]
- Test antioxidants: Compounds or extracts for analysis
- UV-Vis spectrophotometer with 1 cm pathlength cuvettes [110] [111]
- Analytical balance and adjustable micropipettes
Safety Precautions:
- Wear appropriate PPE (gloves, lab coat, eye protection)
- DPPH may cause irritation - minimize skin contact and inhalation [108]
- Methanol/ethanol are flammable - handle away from ignition sources [108]
Procedure:
- DPPH Stock Solution Preparation (0.1 mM):
- Accurately weigh 2 mg DPPH
- Dissolve in 100 mL methanol (final concentration ~0.1 mM)
- Protect from light by wrapping container in aluminum foil [108]
- Store refrigerated until use
Working Solution Verification:
- Dilute stock solution to achieve absorbance ~1.0 ± 0.1 at 517 nm
- Prepare fresh daily due to photosensitivity [108]
Sample Preparation:
- Prepare serial dilutions of test compounds in same solvent
- Include Trolox standard (positive control) at concentrations: 10, 25, 50, 100, 250 μM
- Prepare solvent blank (negative control)
Reaction Setup:
- Pipette 0.1 mL of each sample dilution into separate cuvettes
- Add 3.0 mL DPPH working solution to each cuvette [109]
- Mix thoroughly by inversion or gentle vortexing
- Include negative control (DPPH + solvent)
- Prepare sample background controls (sample + solvent without DPPH)
Incubation and Measurement:
- Incubate reactions in dark at 25°C for 30 minutes [109]
- Measure absorbance at 517 nm against methanol blank
- Record measurements in triplicate for statistical validity
Figure 2: DPPH assay workflow from reagent preparation to data analysis
Data Analysis and Calculations
Scavenging Activity Calculation:
- Calculate percentage radical scavenging activity for each concentration:
% Scavenging = [(Acontrol - Asample) / A_control] × 100 [108]
Where: Acontrol = Absorbance of negative control, Asample = Absorbance of test sample
IC₅₀ Determination:
- Plot % scavenging activity versus sample concentration
- Fit dose-response curve using non-linear regression
- Determine concentration providing 50% scavenging (IC₅₀)
- Lower IC₅₀ indicates higher antioxidant potency [108]
Trolox Equivalent Antioxidant Capacity (TEAC):
- Calculate TEAC values using the formula:
TEAC = IC₅₀ (Trolox) / IC₅₀ (sample) [106]
- TEAC > 1 indicates superior activity to Trolox standard
Table 1: Key Parameters from CV and DPPH Assay Correlation
| CV Parameter | DPPH Parameter | Correlation Relationship | Interpretation in Drug Development |
|---|---|---|---|
| Oxidation Potential (E°) | IC₅₀ Value | Inverse correlation | Lower E° and IC₅₀ indicate higher antioxidant potency |
| Number of Oxidation Waves | TEAC Value | Direct proportionality | Multiple redox centers increase scavenging capacity |
| Peak Current Ratio (ipc/ipa) | Reaction Kinetics | Reversibility indicates stable antioxidant intermediates | Important for sustained antioxidant protection |
| Scan Rate Dependence | Time-course Scavenging | Diffusion-controlled systems show concentration-dependent activity | Predicts bioavailability and tissue penetration |
Research Reagent Solutions
Table 2: Essential Materials for Combined CV-DPPH Antioxidant Studies
| Reagent/Equipment | Function/Specification | Application Notes |
|---|---|---|
| DPPH (2,2-diphenyl-1-picrylhydrazyl) | Stable free radical source (MW = 394.32) | Light-sensitive; prepare fresh solutions daily [108] |
| Trolox Standard | Water-soluble vitamin E analog (Reference antioxidant) | TEAC calibration; typical working range: 10-500 μM [106] |
| Methanol (HPLC grade) | Spectrophotometric solvent for DPPH | Low UV cutoff; minimal antioxidant interference [108] |
| Phosphate Buffer (0.1 M, pH 7.4) | Physiological simulation in CV | Maintains biological relevance for drug development studies |
| Glassy Carbon Electrode | Working electrode for CV | Polish with 0.05 μm alumina suspension between measurements |
| UV-Vis Spectrophotometer | Absorbance measurement at 517 nm | 1 cm pathlength quartz or glass cuvettes [110] |
| Cuvettes | Sample containment for spectrophotometry | Quartz for UV, glass/plastic for visible range [111] |
Advanced Applications and Synergy Studies
Investigating Antioxidant Interactions
The combination of CV and DPPH assays enables sophisticated analysis of antioxidant interactions in complex mixtures:
- Synergistic Effects: CV identifies favorable redox potential matching that enables electron transfer between antioxidants
- Antagonistic Effects: DPPH reveals interference between compounds, such as trehalose inhibition of phenolic antioxidants [112]
- Checkerboard Assay Adaptation: Two-dimensional microdilution for efficient binary mixture analysis [112]
Structure-Activity Relationship (SAR) Studies
Large-scale DPPH screening combined with electrochemical parameters enables robust SAR analysis:
- Phenol Substitution Patterns: Ortho-diphenolic structures (catechols) show enhanced activity due to stabilization of phenoxyl radicals
- Conjugation Effects: Extended π-systems lower oxidation potentials and enhance radical stabilization
- Ionization Potential Correlation: Computational parameters (BDE, IP, PDE) complement experimental CV and DPPH data [106]
Table 3: Correlation Trends for Major Phenolic Antioxidant Classes
| Compound Class | Typical Oxidation Potential (E° vs. Ag/AgCl) | IC₅₀ Range (μM) | Structural Features Enhancing Activity |
|---|---|---|---|
| Simple Phenols | +0.6 to +0.9 V | 50-200 | Multiple hydroxyl groups, electron-donating substituents |
| Catechols | +0.2 to +0.5 V | 10-50 | Ortho-dihydroxy arrangement for radical stabilization |
| Flavonoids | +0.3 to +0.7 V | 5-40 | C2-C3 double bond, 3-OH, 4-oxo, catechol B-ring |
| Hydroxycinnamates | +0.4 to +0.8 V | 15-80 | Conjugated side chain, methoxy substitutions |
Troubleshooting and Technical Considerations
Common Experimental Issues
- DPPH Solution Instability: Absorbance decrease >10% indicates decomposition; prepare fresh solution protected from light [108]
- Non-linear Calibration Curves: Excessive concentration ranges; optimize for linear 20-80% scavenging range
- Sample Interference: Colored compounds or those absorbing at 517 nm require background correction [108]
- Irreversible CV Responses: May indicate follow-up chemical reactions (EC mechanism); use faster scan rates to detect intermediates
- Solvent Compatibility: Ensure DPPH and test compounds are soluble in reaction medium; <5% DMSO is typically acceptable
Method Validation Guidelines
- Precision: Intra-assay CV < 10% for replicate measurements
- Accuracy: Trolox recovery 85-115% in standard addition experiments
- Linearity: R² > 0.98 for calibration curves in working range
- Limit of Detection: Typically 1-5 μM for most phenolic antioxidants
The strategic correlation of cyclic voltammetry with DPPH scavenging assays and spectrophotometry provides a multidimensional approach to antioxidant characterization. CV delivers thermodynamic and electron-transfer parameters, while DPPH assays quantify practical radical scavenging efficiency under controlled conditions. This combined methodology offers drug development researchers a powerful toolkit for rapid screening, mechanism elucidation, and structure-activity relationship studies of novel antioxidant compounds. The standardized protocols presented herein ensure reproducibility across laboratories while maintaining flexibility for specific research applications.
In the broader context of cyclic voltammetry (CV) for redox reaction analysis, the ability to accurately predict redox potentials is fundamental for advancing applications in energy storage, drug development, and synthetic chemistry. Density Functional Theory (DFT) offers a powerful computational approach to model these electrochemical parameters at the atomic level, providing a crucial link between theoretical insights and experimental observations [113] [41]. This protocol details the application of DFT for calculating redox potentials, a key thermodynamic property, and validates these computational findings against experimental cyclic voltammetry data. The integration of the scheme of squares framework allows for a systematic analysis of complex reaction mechanisms involving coupled electron and proton transfers, which are often encountered in redox-active systems for flow batteries and pharmaceutical compounds [113] [114] [41].
Theoretical Background
Redox Potential and the Nernst Equation
In electrochemistry, the redox potential quantifies the tendency of a species to gain or lose electrons. For a reduction process, the standard potential ((E^{0}{ox/red})) relates to the change in Gibbs free energy ((\Delta G)) according to: [ E^{0}{ox/red} = -\frac{\Delta G}{nF} ] where (n) is the number of electrons transferred and (F) is the Faraday constant [41]. In practice, the measured potential is influenced by reactant and product activities, as described by the Nernst equation. When proton transfer accompanies electron transfer, the potential becomes pH-dependent [41].
The Scheme of Squares Framework
The electrochemical scheme of squares is a conceptual model used to disentangle complex redox mechanisms involving multiple electron and proton transfers [41]. It diagrams possible pathways, distinguishing between decoupled electron transfer (ET) and proton transfer (PT), and concerted proton-electron transfer (PET). This framework is critical for interpreting CV data and designing computational studies that accurately reflect the underlying reaction chemistry [41].
Computational Protocols
The following diagram illustrates the logical workflow for computing and validating redox potentials using DFT, from initial setup to final benchmarking.
Detailed Methodology
Geometry Optimization and Energy Calculation
- Software and Tools: Calculations are performed using quantum chemistry software such as Gaussian 16 [41].
- Functional and Basis Set: Employ the M06-2X functional with the 6-31G(d) basis set for geometry optimization and frequency analysis. For higher accuracy single-point energy calculations, use a larger basis set like Def2-TZVP [41].
- Solvation Model: Incorporate an implicit solvation model, such as SMD (Solvation Model based on Density) or CPCM-X (Extended Conductor-like Polarizable Continuum Model), during optimization and energy calculations to account for solvent effects [115] [41].
- System Preparation: Optimize the geometries of both the oxidized and reduced states of the molecule. For proton-coupled electron transfer, optimize the relevant protonation states.
Frequency Analysis
- Perform frequency calculations at the same level of theory as the geometry optimization to confirm the identification of true local minima (no imaginary frequencies) and to obtain thermochemical corrections, including zero-point vibrational energy and thermal corrections to enthalpy and entropy, for calculating Gibbs free energy [41].
Redox Potential Calculation
- Calculate the change in Gibbs free energy ((\Delta G)) for the reduction (or oxidation) process. For electron transfer only: ( \text{X}^{m+} + n e^- \rightarrow \text{X}^{(m-n)+} ). For concerted proton-electron transfer: ( \text{X}^{m+} + n e^- + np\text{H}^+ \rightarrow \text{XH}{n_p}^{(m-n)+} ) [41].
- Compute the standard redox potential ((E^{0})) using the equation: [ E^{0}{ox/red} = -\frac{\Delta G}{nF} - E{\text{SHE}} ] where (E_{\text{SHE}}) is the potential of the standard hydrogen electrode (typically 4.28 V in the computational standard hydrogen electrode framework) [41].
Calibration with Experimental Data
- Calibrate computed redox potentials against experimental CV data to correct for systematic errors inherent in the DFT functionals and solvation models. This is achieved by applying a linear scaling factor derived from a set of reference molecules with known experimental potentials [113] [41].
Benchmarking and Validation
Performance of Computational Methods
Quantitative benchmarking against experimental datasets is crucial for assessing the predictive accuracy of computational methods. The table below summarizes the performance of various methods, including DFT functionals and neural network potentials (NNPs), in predicting reduction potentials for main-group and organometallic species [115].
Table 1: Benchmarking of Computational Methods for Reduction Potential Prediction
| Method | System Type | Mean Absolute Error (MAE/V) | Root Mean Squared Error (RMSE/V) | Coefficient of Determination (R²) |
|---|---|---|---|---|
| B97-3c (DFT) | Main-Group (OROP) | 0.260 | 0.366 | 0.943 |
| Organometallic (OMROP) | 0.414 | 0.520 | 0.800 | |
| GFN2-xTB (SQM) | Main-Group (OROP) | 0.303 | 0.407 | 0.940 |
| Organometallic (OMROP) | 0.733 | 0.938 | 0.528 | |
| UMA-S (NNP) | Main-Group (OROP) | 0.261 | 0.596 | 0.878 |
| Organometallic (OMROP) | 0.262 | 0.375 | 0.896 |
SQM: Semiempirical Quantum Mechanical; NNP: Neural Network Potential. Data adapted from VanZanten & Wagen (2025) [115].
Analysis of Benchmarking Data
- DFT Performance: The B97-3c functional demonstrates strong predictive power for main-group organic molecules (MAE ~0.26 V) but shows increased error for organometallic complexes (MAE ~0.41 V) [115].
- Semiempirical Methods: GFN2-xTB provides a fast alternative with reasonable accuracy for main-group systems (MAE ~0.30 V), but its performance significantly degrades for organometallics (MAE ~0.73 V) [115] [116].
- Emerging NNPs: Neural Network Potentials like UMA-S show promise, achieving accuracy comparable to DFT for organometallic species (MAE ~0.26 V) while being less accurate for main-group molecules in some cases [115].
- Practical Considerations: The relatively low sensitivity of errors to the level of theory (with MAEs typically around 0.2-0.3 V) suggests that faster, lower-cost methods can be pragmatically chosen for high-throughput screening, reserving more expensive DFT calculations for final validation [116].
The Scientist's Toolkit
Table 2: Essential Research Reagent Solutions and Computational Tools
| Tool/Reagent | Function/Description | Example Use/Note |
|---|---|---|
| Gaussian 16 | Quantum chemistry software package. | Used for DFT calculations, geometry optimization, and frequency analysis [41]. |
| Psi4 | Open-source quantum chemistry package. | Used for DFT computations, supports various functionals and solvation models [115]. |
| B97-3c Functional | A composite DFT functional. | Provides good accuracy for redox potentials of organic molecules [115]. |
| M06-2X Functional | A hybrid meta-GGA DFT functional. | Known for reliable reaction energies of organic transformations [41]. |
| SMD Model | Implicit solvation model. | Accounts for solvent effects in energy calculations [41]. |
| CPCM-X | Implicit solvation model. | An extended conductor-like model for improved solvation free energies [115] [116]. |
| Def2-TZVP | Triple-zeta valence basis set. | Used for accurate single-point energy calculations [41]. |
| Standard Hydrogen Electrode (SHE) | Computational reference electrode. | Essential for reporting computed potentials against a standard scale [41]. |
Application Notes
- Interpreting CV with Computation: DFT-calculated redox potentials and the scheme of squares can directly aid in interpreting cyclic voltammograms by identifying the number of oxidation states, their stability, and whether irreversibility arises from coupled chemical reactions like proton transfer [41].
- Drug Development Context: In pharmaceutical sciences, understanding redox properties is critical for predicting drug metabolism and stability. DFT can model the electronic structure of chemotherapeutic drugs, and when combined with QSPR analysis, can help predict thermodynamic properties and biological activity [117] [114].
- Limitations and Error Sources: Key challenges include accurately modeling charged systems and deficiencies in exchange-correlation functionals, leading to systematic errors. Implicit solvation models may not fully capture specific solvent-solute interactions. Calibration against experimental data is, therefore, highly recommended [41].
This application note establishes a validated protocol for using DFT to model and predict redox potentials, directly supporting research that integrates cyclic voltammetry with computational analysis. The structured workflow, benchmarking data, and essential toolkit provide researchers with a practical guide for implementing these methods. By bridging computational insights with experimental validation, this approach deepens the understanding of redox mechanisms and enhances the predictive design of new molecular systems for energy storage and medicine.
The Electrochemical 'Scheme of Squares' for Elucidating Complex ET/PT (Electron/Proton Transfer) Mechanisms
The study of electron transfer (ET) and proton transfer (PT) is fundamental to understanding mechanisms in diverse fields ranging from energy storage to biological systems and drug development [41]. These reactions rarely occur in isolation; rather, they often proceed through coupled mechanisms that can be challenging to decipher using experimental techniques alone. The electrochemical "Scheme of Squares" (also referred to as the "square scheme") provides a powerful theoretical framework for diagramming these complex proton-coupled electron transfer (PCET) pathways [41] [118]. This framework systematically represents possible sequences of decoupled ET and PT steps along the sides of a square, or coupled PET steps along its diagonal [41].
When combined with cyclic voltammetry (CV)—a technique that measures current response to applied potential—the Scheme of Squares enables researchers to interpret redox mechanisms, determine thermodynamic parameters, and assess electrochemical reversibility [41] [50]. For researchers in drug development, understanding these mechanisms is crucial for studying the redox behavior of pharmaceutical compounds, their stability, and their metabolic pathways [41] [50]. This Application Note provides detailed protocols for implementing the Scheme of Squares framework to elucidate complex ET/PT mechanisms, with a focus on practical computational and experimental methodologies.
Theoretical Framework
Fundamentals of the Scheme of Squares
The Scheme of Squares provides a systematic method for representing and analyzing the possible pathways in proton-coupled electron transfer reactions. For a one-electron, one-proton process, the framework diagrams four distinct species at the corners of a square connected by horizontal ET reactions and vertical PT reactions [41] [118]. The overall reaction from the fully oxidized to fully reduced species can proceed along multiple pathways: sequential ET followed by PT, sequential PT followed by ET, or a concerted PET process along the diagonal [41].
The thermodynamic driving forces for these reactions are governed by the Nernst equation for electron transfers and by acid dissociation constants (pKa) for proton transfers [41]. For a reversible system, the formal potential ((E^0_{ox/red})) can be computed from the change in Gibbs free energy ((\Delta G)) using the equation [41]:
[ E^0_{ox/red} = -\frac{\Delta G}{nF} ]
where (n) is the number of electrons transferred, and (F) is the Faraday constant.
When proton transfers are coupled to electron transfers, the solution pH significantly influences the observed formal potential. The generalized Nernst equation for such systems becomes [41]:
[ E = E^0{ox/red} - \frac{RT}{nF} \ln \left( \frac{a{red}}{a_{ox}} \right) - \frac{RT \ln(10)}{F} \cdot \text{pH} ]
where (R) is the universal gas constant, (T) is temperature, and (a{red}) and (a{ox}) represent the activities of reduced and oxidized species, respectively.
Table 1: Key Thermodynamic Parameters in Scheme of Squares Analysis
| Parameter | Symbol | Description | Experimental Determination |
|---|---|---|---|
| Formal Potential | (E^0) | Standard potential for redox couple | Cyclic Voltammetry |
| Acid Dissociation Constant | pKa | Measure of acid strength | Potentiometric titration |
| Bond Dissociation Free Energy | BDFE | Free energy to break X-H bond | Calorimetry, computational chemistry |
| Potential of Hydrogenation | E°(V vs H₂) | Potential for H₂ addition to a species | Electrochemical measurement |
Diagnostic Criteria for Reaction Pathways
The Scheme of Squares enables distinction between different PCET mechanisms through analysis of Pourbaix diagrams (plots of formal potential versus pH) [118]. The slopes of these diagrams reveal the proton-to-electron stoichiometry:
- A slope of approximately -59 mV/pH at 25°C suggests a 1e⁻/1H⁺ transfer process
- A slope of approximately -118 mV/pH indicates a 2e⁻/2H⁺ transfer process
- A zero slope indicates a pH-independent electron transfer process
The concept of "crossed potentials"—where the apparent reduction potential for the second electron transfer ((E^{0}{app2})) is more positive than that for the first ((E^{0}{app1}))—can lead to stabilization of intermediate states and influence the preferred reaction pathway [118]. This potential inversion often occurs when the one-electron reduced species is a stronger base than the two-electron reduced species (pKa₂ > pKa₁), which can be identified through careful analysis of the pH dependence of formal potentials [118].
Computational Protocols
Density Functional Theory (DFT) Calculations
Purpose: To calculate thermodynamic parameters (redox potentials, pKa values, and reaction energies) for constructing Scheme of Squares diagrams and predicting PCET pathways.
Workflow:
Molecular Geometry Optimization
Single-Point Energy Calculations
Thermodynamic Parameter Calculation
- Compute redox potentials using the equation: (E^0 = -\Delta G/nF)
- Calculate pKa values from the free energy difference between protonated and deprotonated forms
- Determine bond dissociation free energies (BDFEs) for X-H bonds [119]
Table 2: Computational Settings for DFT Calculations
| Calculation Step | Method | Basis Set | Solvation Model | Software Reference |
|---|---|---|---|---|
| Initial Optimization | PM7 | N/A | SMD | Gaussian 16 [41] |
| Higher Optimization | M06-2X | 6-31G(d) | SMD | Gaussian 16 [41] |
| Single-Point Energy | M06-2X | Def2-TZVP | SMD | Gaussian 16 [41] |
| Frequency Calculation | M06-2X | 6-31G(d) | SMD | Gaussian 16 [41] |
Calibration to Experimental Data
Purpose: To improve the accuracy of calculated thermodynamic parameters by scaling them to experimental benchmarks.
Procedure:
Select Reference Compounds
- Choose molecules with reliable experimental redox potentials and pKa values
- Ensure structural diversity to cover the chemical space of interest
- Include compounds from the field of redox flow batteries, which provide well-characterized reference data [41]
Perform Scaling
- Calculate scaling factors by regression of computed values against experimental data
- Apply separate scaling factors for redox potentials and pKa values
- For redox potentials, accuracy of approximately 0.1 V can be achieved after calibration [41]
Validate the Model
- Test scaled parameters on molecules not included in the training set
- Verify that the calibrated model reproduces experimental cyclic voltammograms
Experimental Protocols
Cyclic Voltammetry for PCET Analysis
Purpose: To experimentally characterize PCET reactions and validate computational predictions.
Materials and Equipment:
- Potentiostat with capability for cyclic voltammetry measurements [50]
- Electrochemical cell with three-electrode configuration:
- Working electrode (glassy carbon, platinum, or gold)
- Counter electrode (platinum wire or mesh)
- Reference electrode (Ag/AgCl, SCE, or Fc/Fc⁺ for non-aqueous systems)
- Electrolyte solution with appropriate supporting electrolyte
- Analyte dissolved in suitable solvent
- pH buffer solutions for studies as a function of pH
Procedure:
Solution Preparation
- Prepare electrolyte solution with supporting electrolyte (e.g., 0.1 M TBAPF₆ for non-aqueous systems)
- Dissolve analyte to appropriate concentration (typically 1-5 mM)
- For pH-dependent studies, prepare series of buffered solutions covering relevant pH range
Instrument Setup
- Purge solution with inert gas (N₂ or Ar) for 10-15 minutes to remove oxygen
- Set initial and final potentials based on solvent electrochemical window [41]
- Select appropriate scan rate (typically 0.01-1 V/s)
- For reversible systems, use multiple scan rates to study kinetics
Data Collection
- Record cyclic voltammograms at multiple pH values
- Perform experiments at different scan rates to distinguish diffusion-controlled and surface-bound processes
- Collect blank voltammograms of electrolyte without analyte for background subtraction
Data Analysis
- Determine formal potentials from the average of anodic and cathodic peak potentials for reversible systems [50]
- Calculate peak separation ((ΔEp = E{pa} - E_{pc})) to assess electrochemical reversibility
- Construct Pourbaix diagram by plotting formal potential versus pH
- Analyze peak current dependence on scan rate to determine if species is diffusion-controlled or surface-bound
Table 3: Troubleshooting Common CV Issues
| Problem | Possible Cause | Solution |
|---|---|---|
| Large peak separation (>59 mV) | Slow electron transfer kinetics | Use slower scan rate or check electrode surface |
| No redox peaks | Analyte concentration too low | Increase concentration or check solubility |
| Drifting baseline | Uncompensated resistance | Use lower concentration of supporting electrolyte |
| Irreversible waves | Chemical reaction following ET | Vary scan rate to study follow-up kinetics |
Analysis of CV Data for Scheme of Squares
Purpose: To extract thermodynamic parameters from CV data and construct Scheme of Squares diagrams.
Procedure:
Determine Formal Potentials
- For reversible systems: (E^0 = (E{pa} + E{pc})/2) [50]
- For quasi-reversible systems: use digital simulation to extract formal potentials
Identify Proton-Coupled Transitions
- Plot formal potentials versus pH to create Pourbaix diagram
- Identify break points where mechanism changes
- Calculate proton-to-electron ratio from slopes
Assess Electrochemical Reversibility
Construct Scheme of Squares
- Place experimental species at corners of square
- Label edges with calculated ET potentials and PT pKa values
- Identify dominant pathways based on thermodynamic parameters
The Scientist's Toolkit
Essential Research Reagent Solutions
Table 4: Key Reagents and Materials for Scheme of Squares Studies
| Reagent/Material | Function | Example Specifications | Application Notes |
|---|---|---|---|
| Supporting Electrolytes | Provide ionic conductivity | TBAPF₆ (0.1 M), KCl (0.1 M) | Use non-coordinating ions for non-aqueous systems |
| Buffers | Control pH in aqueous systems | Phosphate, acetate, Britton-Robinson | Use at appropriate concentration (0.05-0.1 M) |
| Internal Standards | Reference potentials | Ferrocene/Ferrocenium (Fc/Fc⁺) | Add after measurements for non-aqueous CV |
| Electrode Polishing | Maintain reproducible surface | Alumina slurry (0.3, 0.05 μm) | Polish electrode before each experiment |
| Solvents | Dissolve analytes | Acetonitrile, DMSO, water | Use HPLC grade; dry non-aqueous solvents |
Software and Computational Tools
- Gaussian 16: For DFT calculations including geometry optimizations and frequency calculations [41]
- SMD Solvation Model: For implicit solvation in computational chemistry [41]
- Digital Simulation Software: For fitting CV data to reaction mechanisms (commercial or custom-coded)
- Bayesian Fitting Frameworks: For parameter estimation from voltammetric data [118]
Case Study: Disulfide Bond Redox Chemistry
To illustrate the practical application of the Scheme of Squares framework, we examine the disulfide bond reductive cleavage and oxidative formation in Escherichia coli hydrogenase maturation factor HypD, which represents a net two-proton, two-electron transfer process [118].
Experimental Conditions:
- Protein immobilized on graphite electrode
- pH range: 4.0 to 9.0
- Technique: Fourier transformed alternating current voltammetry (FTACV)
- Data analysis: Automated fitting within Bayesian framework [118]
Key Findings:
- Across the entire pH range studied, the data was best modeled by two separate, stepwise one-electron, one-proton transfers
- The apparent reduction potential for the second transfer ((E^{0}{app2})) was approximately 10 mV more positive than for the first transfer ((E^{0}{app1})) at pH 6.0 [118]
- This small but significant potential crossover enables stabilization of the one-electron reduced intermediate
- The stepwise mechanism prevailed even under extreme pH conditions where a concerted two-electron transfer might be expected
Interpretation: The persistence of the stepwise mechanism across pH conditions highlights the importance of analyzing PCET reactions over a wide pH range to accurately determine mechanism. The "crossed" potentials ((E^{0}{app2} > E^{0}{app1})) result from the one-electron reduced species being a stronger base than the two-electron reduced species (pKa₂ > pKa₁), which is a common feature in biological redox systems [118].
Advanced Applications
Analyzing Multi-Electron, Multi-Proton Transitions
For systems involving multiple electrons and protons, the basic Scheme of Squares expands into more complex cubic or hypercube schemes. The same fundamental principles apply, but the analysis requires careful consideration of all possible intermediates. The use of FTACV enhances the ability to resolve these complex mechanisms by emphasizing fast electron transfer processes through higher harmonic components [118].
Pharmaceutical Applications
In drug development, the Scheme of Squares framework can be applied to:
- Study metabolic oxidation/reduction pathways of drug candidates
- Predict redox-induced toxicity mechanisms
- Design prodrugs with specific activation mechanisms
- Understand the redox behavior of pharmacophores with quinone, hydroquinone, or other redox-active moieties
The combination of computational prediction with experimental validation through CV provides a powerful approach for screening compound libraries for undesirable redox behavior early in the drug development process.
In the field of electrochemistry, particularly in cyclic voltammetry (CV) for redox reaction analysis, a significant challenge is the selective identification and quantification of specific analytes within complex mixtures. Cyclic voltammetry is a powerful technique that investigates the kinetics and mechanisms of electrochemical reactions by applying a linearly varying potential to an electrode surface and monitoring the corresponding current response, resulting in a current-potential (i-E) plot [120]. However, voltammetric signals from complex biological or environmental samples often overlap, making it difficult to resolve individual contributors.
Principal Components Regression (PCR) addresses this challenge by combining the data-reduction capabilities of Principal Component Analysis (PCA) with inverse least-squares regression [121]. This multivariate data analysis approach is routinely used to predict neurochemical concentrations from in vivo fast-scan cyclic voltammetry measurements and can be rapidly employed with present-day computer programming languages [121]. For researchers in drug development working with complex electrochemical data, PCR provides a robust mathematical framework for extracting meaningful, selective information about target analytes that would otherwise remain obscured in overlapping voltammetric signals.
Theoretical Foundation: PCR in Electrochemical Analysis
Relationship Between PCA, PCR, and PLS-Regression
PCR belongs to a family of multivariate statistical techniques that includes Principal Component Analysis (PCA) and Partial Least Squares Regression (PLSR). Understanding their relationships is crucial for selecting the appropriate method for analyte identification:
PCA is an unsupervised technique that decomposes the data matrix into a new set of uncorrelated variables (Principal Components) that explain the maximum amount of variance in the data set [122]. While versatile for data exploration, PCA does not directly model the relationship between measurements and concentrations.
PCR builds upon PCA by using the principal components as independent variables in a regression model calibrated to reference concentration values [121]. This two-stage process first reduces data dimensionality then establishes predictive relationships.
PLSR is a supervised method that simultaneously models the relationship between two matrices (the sensor array measurements and class affiliation matrix) while performing dimensionality reduction [122] [123]. Unlike PCR, PLSR considers the covariance between measurement data and target variables during decomposition.
For process monitoring and output prediction applications, PLSR often demonstrates advantages, particularly when quality variables are affected by process conditions [123]. However, PCR remains valuable for latent pattern analysis in datasets and provides more interpretable components in certain electrochemical applications.
Mathematical Framework of PCR
In PCR, a training set containing reference voltammograms at known concentrations is assembled. Abstract representations of the training set voltammograms called principal components (PCs) are calculated, with PCs describing relevant information retained and those describing noise discarded [121]. The projection of the training set voltammograms onto the relevant PCs (called scores) are calibrated to the reference concentration values through regression analysis.
The PCR prediction of unknown neurochemical concentrations (Cunk) can be described according to:
where Aunk contains the unknown cyclic voltammograms to be predicted, Vc contains the relevant PCs of rank r, and F contains the regression coefficients that relate unknown concentrations of each analyte to the scores of the relevant PCs [121].
Table 1: Key Mathematical Components in PCR Analysis
| Component | Symbol | Description | Role in Analysis |
|---|---|---|---|
| Training Set Voltammograms | ATS | n × m matrix of reference measurements | Provides baseline data for model building |
| Principal Components | Vc | Abstract representations of training set | Captures essential variance while reducing noise |
| Regression Coefficients | F | Matrix relating PC scores to concentrations | Enables prediction of unknown samples |
| Regression Vector | kj | Cyclic voltammetric representation for analyte j | Qualitative assessment of model appropriateness |
Each column of the K matrix (pseudoinverse of FVCT), kj, can be thought of as a cyclic voltammetric representation of the regression vector for each analyte in the relevant multivariate calibration space of the training set [121]. This vector represents the PCR interpretation of sensitivity at each potential for a specific analyte j based on the training set voltammograms, reference concentration values, and the relevant PCs of the multivariate model.
Experimental Design and Workflow
The following diagram illustrates the complete PCR workflow for analyte identification and quantification from cyclic voltammetry data:
Data Collection Parameters for Cyclic Voltammetry
Proper experimental design is crucial for generating high-quality data for PCR analysis. The table below summarizes key parameters for cyclic voltammetry data collection:
Table 2: Cyclic Voltammetry Parameters for PCR Analysis
| Parameter | Typical Range | Considerations for PCR | Impact on Data Quality |
|---|---|---|---|
| Scan Rate | 0.01-5 V/s (standard studies) [120] | Affects peak separation and current response | Higher rates increase current but may cause incomplete reactions |
| Potential Range | ±2.0 V (aqueous) to ±5.0 V (organic) [120] | Must encompass all redox events of interest | Insufficient range may miss relevant electrochemical processes |
| Quiet Time | 5-60 seconds [120] | Allows electrode stabilization | Reduces baseline drift and improves reproducibility |
| Cycle Number | 3-50 repetitions [120] | Provides data for assessing reproducibility | Multiple cycles enable signal averaging and noise reduction |
| Data Points | 2000 per cycle [120] | Affects resolution of voltammetric features | Higher density improves definition of peak shapes |
Training Set Design Considerations
The training set is the foundation of a robust PCR model and should include voltammograms at known concentrations that encompass the expected variation in future unknown samples. Key considerations include:
- Concentration Range: The training set should adequately cover the concentration range expected in unknown samples, with standards at both low and high concentrations.
- Matrix Effects: Include samples with varying background compositions that mimic real samples to account for matrix effects.
- Number of Standards: Traditionally, five cyclic voltammograms per analyte are incorporated into a training set, though this may vary based on complexity [121].
- Leverage Assessment: Samples with high leverage (hi > 3r/m, where r is number of PCs and m is number of samples) may need evaluation, though strict exclusion is not always practical for in vivo FSCV training sets [121].
Computational Protocol for PCR Implementation
Step-by-Step PCR Analysis Procedure
Data Preprocessing
- Arrange training set voltammograms in matrix ATS (size n × m, where n is potential steps and m is number of samples)
- Arrange reference concentration values in matrix CTS (size j × m, where j is number of analytes)
- Apply preprocessing if needed (mean-centering, scaling) based on data characteristics
Principal Component Analysis
- Perform singular value decomposition on ATS to extract principal components
- Determine optimal number of components (r) to retain using scree plots, cross-validation, or residual analysis
- Select Vc containing the relevant PCs of rank r
Regression Model Development
- Calculate projection of training set onto relevant PCs: AprojTS = ATST · Vc
- Compute regression coefficients: F = (AprojTS · AprojTST)-1 · AprojTS · CTST [121]
Model Validation
Prediction of Unknown Samples
- Project unknown voltammograms (Aunk) onto relevant PCs: Scoresunk = Aunk · Vc
- Predict concentrations: Cunk = Scoresunk · F
Model Interpretation
- Calculate K matrix (pseudoinverse of FVCT) to obtain cyclic voltammetric representations of regression vectors
- Examine shape of each kj vector to assess chemical appropriateness of the model [121]
Diagnostic Tools for Model Assessment
Several diagnostic tools can evaluate and improve multivariate calibration models:
- Leverage (hi): Measures how far a sample is from other training set samples in the calibration space, with values between 0 and 1 [121]
- Studentized Residual (ti): Standardized difference between estimated and reference concentration values [121]
- Cook's Distance (Di): Combines leverage and studentized residual to measure the effect of the ith sample on the overall multivariate calibration [121]
- Residual Color Plots: Visual tool for interpreting model residuals and identifying patterns
- K Matrix Analysis: Qualitative evaluation of pure analyte cyclic voltammograms determined from the PCR calibration relationship [121]
Research Reagent Solutions and Materials
Table 3: Essential Materials for PCR-Based Electrochemical Analysis
| Material/Reagent | Specifications | Function in Analysis |
|---|---|---|
| Glassy Carbon Electrode | 3 mm diameter, polished with 0.3 μm alumina [96] | Standard working electrode for voltammetric measurements |
| Reference Electrode | Ag/AgCl or Saturated Calomel (SCE) [96] [124] | Provides stable potential reference during scans |
| Counter Electrode | Platinum wire [96] | Completes electrical circuit without interference |
| Supporting Electrolyte | Phosphate buffer, carbonate buffer, or [n-Bu4N][PF6] in organic systems [96] [28] | Provides ionic conductivity without participating in redox reactions |
| Standard Solutions | Analytical grade analytes at known concentrations [121] | Creates training set for multivariate calibration |
| Data Acquisition System | Potentiostat with multiscan capability (1×10⁻⁴ to 10,000 V/s) [120] | Controls potential application and current measurement |
| Computational Software | MATLAB, R, or Python with linear algebra capabilities [121] | Implements PCR algorithm and statistical diagnostics |
Case Study: PCR for Discrimination of Schisandra Fruits
A practical application of multivariate analysis in electrochemistry demonstrates the utility of these approaches. In a study discriminating between Schisandrae Sphenantherae Fructus (SSF) and Schisandrae Chinensis Fructus (SCF), voltammetric data combined with multivariate analysis successfully differentiated between visually similar fruits [124].
Experimental Protocol
- Sample Preparation: 0.75 g of Schisandra fruit samples were used to prepare methanol extracts in 10 mL solvent [124]
- Voltammetric Analysis: Potential sweep from 0 V to +1.0 V vs. Ag/AgCl at 0.02 V/s sampling interval [124]
- Data Collection: Oxidation current values at 250 points between +0.5 V to +1.0 V were recorded [124]
- Multivariate Analysis: Data sets of oxidation current values were applied to PLS-DA (a related technique to PCR) for discrimination [124]
Results and Interpretation
The PCA of voltammetric data showed variances of 54.6% (PC1) and 36.4% (PC2), with cumulative variance of 91.0% [124]. The scores for SSF samples clustered in different quadrants than SCF samples, enabling clear discrimination. This case study demonstrates how electrochemical fingerprinting combined with multivariate analysis can successfully distinguish complex samples, illustrating the power of these approaches for analyte identification in pharmaceutical quality control.
Troubleshooting and Optimization Guidelines
Common Issues and Solutions
- Poor Prediction Accuracy: Evaluate leverage and studentized residuals to identify outliers; consider expanding training set to cover concentration range more evenly
- Model Overfitting: Use cross-validation to determine optimal number of principal components; avoid retaining PCs that primarily describe noise
- Electrode Drift Effects: For continuous measurements, analyze smaller epochs separately rather than concatenating results, as electrode drift causes substantial error in predicted concentrations [121]
- Peak Shift Problems: Interpret residual color plots to identify peak shifts and their effect on predicted concentrations [121]
Advantages and Limitations
PCR offers several advantages for electrochemical analysis:
- Handles collinear variables effectively
- Reduces dimensionality while retaining relevant information
- Provides both qualitative and quantitative information about electrode processes
- Rapid implementation with modern computational software
However, limitations include:
- Requires careful training set design
- Sensitive to outliers without proper diagnostics
- Model may need recalibration for new experimental conditions
- Interpretation requires understanding of multivariate statistics
For researchers in drug development applying cyclic voltammetry, PCR provides a powerful tool for extracting selective information about target analytes from complex voltammetric data, enabling more informed decisions in pharmaceutical development and quality control.
Electroanalysis has emerged as a critical tool in the pharmaceutical industry, offering versatile and sensitive methods for drug analysis [125]. Among these techniques, cyclic voltammetry (CV) is a cornerstone for investigating redox reactions and establishing key analytical figures of merit for pharmaceutical compounds [126] [127]. This protocol details the application of CV for quantifying the sensitivity, selectivity, and detection limits of drug analytes, with a specific case study on cocaine detection in saliva [128]. The methodology is designed to provide researchers with a robust framework for electrochemical characterization within drug development and forensic analysis.
Theoretical Framework of Cyclic Voltammetry
Cyclic voltammetry is a potentiodynamic electrochemical measurement technique that provides rich qualitative and quantitative information about electroactive species [127]. In CV, the current at a working electrode is measured while the potential between the working electrode and a reference electrode is swept linearly in time between two set values, known as the vertex potentials [126] [127].
Key Parameters for Analytical Figures of Merit
The analytical utility of CV stems from the relationship between the voltammogram's characteristics and the analyte's properties. For a reversible redox reaction, the peak current ((ip)) is described by the Randles-Ševčík equation [126] [11]: [ip = (2.69 \times 10^5) n^{3/2} A D^{1/2} \nu^{1/2} C_A] where:
- (n) is the number of electrons transferred
- (A) is the electrode area (cm²)
- (D) is the diffusion coefficient (cm²/s)
- (\nu) is the scan rate (V/s)
- (C_A) is the analyte concentration (mol/cm³)
The peak potential ((Ep)) and the separation between anodic and cathodic peak potentials ((\Delta Ep)) provide insights into the redox thermodynamics and kinetics, which are crucial for assessing selectivity [11]. A well-behaved, reversible system exhibits a (\Delta E_p) of approximately (59/n) mV at 25°C [11].
Experimental Protocols
Sensor Modification and Preparation
This protocol utilizes a cocaine-modified carbon screen-printed electrode (SPE) as described by Cardoso et al. [128], adaptable for other drug analytes with inherent electroactivity.
Materials and Reagents
Table 1: Essential Research Reagent Solutions
| Reagent/Material | Function/Application | Specifications |
|---|---|---|
| Screen-Printed Electrodes (SPEs) | Working platform; carbon-based working electrode (3 mm diameter), silver reference, carbon counter electrode [128] | |
| Cocaine Hydrochloride | Primary analyte and electrode modifier [128] | Analytical standard |
| PBS Buffer (pH ~7.4) | Supporting electrolyte and dilution medium [128] | 0.1 M |
| Human Saliva | Complex matrix for real-world sample analysis [128] | Collected from healthy donors |
| Gold Nanoparticles (AuNPs) | Signal amplification and confirmation testing [128] | Conjugated with specific antibodies |
| PalmSens 4 Potentiostat | Instrument for applying potential and measuring current [128] |
Electrode Modification Procedure
- Electrode Pre-treatment: Rinse the SPE with Milli-Q water and air-dry. Dispense 100 µL of PBS buffer onto the electrode and perform square wave voltammetry (SWV) from 0 to 1.5 V with a frequency of 15 Hz, amplitude of 25 mV, and step potential of 5 mV. Repeat this pre-treatment three times [128].
- Analyte Modification: Prepare a deposition solution (COCi) of cocaine hydrochloride in PBS at the desired modification concentration. Drop-cast a precise volume (e.g., 100 µL) of this solution onto the working electrode surface [128].
- Drying and Storage: Air-dry the modified electrodes for approximately six minutes. Store the modified electrodes in a sealed bag containing an oxygen adsorbent until use to preserve stability [128].
Cyclic Voltammetry Measurement Protocol
- Instrument Setup: Connect the potentiostat to the computer and initialize the measurement software (e.g., PSTrace) [128].
- Solution Preparation: Prepare a series of standard solutions of the target drug (e.g., cocaine) in PBS buffer and in filtered human saliva, covering the concentration range of interest (e.g., 0 to 1000 ng mL⁻¹) [128].
- Electrochemical Cell Assembly: Place the modified SPE in the electrochemical cell containing the sample solution. Ensure electrical contacts are secure.
- Parameter Configuration: Set the CV parameters in the software:
- Data Acquisition: Run the CV measurement. The system will record the current response as the potential is swept forward and backward, generating the voltammogram.
- Replication: Perform all measurements in triplicate to ensure statistical significance.
Data Analysis Workflow
The following pathway outlines the core data processing steps to establish figures of merit from raw CV data.
- Peak Identification: Identify the oxidation and reduction peak currents ((i{pa}), (i{pc})) and corresponding peak potentials ((E{pa}), (E{pc})) from the voltammogram [11].
- Parameter Extraction: For each concentration, record the peak current and potential values. Calculate the peak potential separation ((\Delta Ep = E{pa} - E{pc})) and the peak current ratio ((i{pa}/i_{pc})) [11].
- Calibration Curve: Plot the peak current ((i_p)) against the analyte concentration for both buffer and saliva matrices. Perform linear regression analysis to obtain the slope (sensitivity), y-intercept, and correlation coefficient (R²) [128].
- Detection and Quantification Limits: Calculate the Limit of Detection (LOD) and Limit of Quantification (LOQ) using the standard deviation of the blank response (σ) and the slope of the calibration curve (S): (LOD = 3.3\sigma/S) and (LOQ = 10\sigma/S) [128].
- Machine Learning for Complex Matrices: To overcome matrix effects in saliva, employ machine learning algorithms (e.g., using Python's scikit-learn or similar) to analyze the full voltammetric response rather than just the peak current, improving accuracy in complex media [128].
Case Study: Cocaine Detection in Saliva
Experimental Workflow
The end-to-end process for detecting an analyte in a complex matrix like saliva involves sample preparation, electrochemical measurement, and advanced data analysis.
Performance Metrics and Data
The following table summarizes the quantitative figures of merit obtained for cocaine detection using the described protocol [128].
Table 2: Analytical Figures of Merit for Cocaine Detection via Cyclic Voltammetry
| Parameter | Value in PBS Buffer | Value in Human Saliva | Method of Determination |
|---|---|---|---|
| Limit of Detection (LOD) | 1.73 ng mL⁻¹ | Not explicitly stated, but successful detection from 0-50 ng mL⁻¹ | Calibration curve (3.3σ/S) [128] |
| Linear Range | Up to 1000 ng mL⁻¹ | 0 to >50 ng mL⁻¹ | Calibration curve linearity [128] |
| Accuracy | N/A | 85% | Machine learning classification vs. actual concentration [128] |
| Assay Time | < 1 minute | < 1 minute | Total measurement time per sample [128] |
| Key Challenge | Minimal | Saliva-to-saliva variation and interference | Addressed via machine learning data processing [128] |
Discussion
The data demonstrates the high sensitivity of the CV-based method, with a LOD of 1.73 ng mL⁻¹ in buffer, which is significantly below the SAMHSA threshold of 20 ng mL⁻¹ for cocaine in saliva [128]. The success in saliva, a complex matrix, highlights the method's robustness. The integration of machine learning is a critical advancement, allowing the sensor to maintain 85% classification accuracy despite inherent biological variations [128]. This approach overcomes a major limitation of traditional electroanalysis in complex media.
The selectivity of the biomolecule-free sensor originates from the specific electroactive characteristics of the cocaine molecule itself, which was confirmed through successful detection in the presence of various interferences like levamisole and caffeine [128]. This combination of intrinsic analyte electroactivity and advanced data analysis provides a powerful tool for rapid, onsite drug testing.
This application note provides a validated protocol for establishing the critical analytical figures of merit—sensitivity, selectivity, and detection limits—for drug analysis using cyclic voltammetry. The case study on cocaine detection proves the method's applicability for rapid, sensitive, and accurate analysis in complex biological matrices. The integration of machine learning for data interpretation presents a modern solution to classical challenges in electroanalysis, paving the way for the development of reliable roadside detection kits and other point-of-care diagnostic tools.
Criteria for Electrochemical Reversibility and Stability Assessment in Flow Battery Molecules
Redox flow batteries (RFBs) represent a pivotal technology for large-scale stationary energy storage, offering scalability, long cycle life, and the ability to decouple power and energy ratings [129]. The efficient operation and commercial viability of RFBs hinge on the performance of their redox-active materials. For organic species in particular, assessing electrochemical reversibility—the ability to undergo repeated redox cycles without degradation—and long-term stability is critical for developing reliable systems [130]. Cyclic voltammetry (CV) serves as a fundamental analytical technique for probing these characteristics at the molecular level, providing invaluable insights into redox potentials, electron transfer kinetics, and degradation mechanisms under controlled conditions [19]. This protocol details standardized methodologies for employing CV to evaluate key performance parameters of redox-active molecules for flow battery applications.
Fundamental Principles of Cyclic Voltammetry
Cyclic voltammetry involves applying a linearly changing potential to an electrochemical cell and measuring the resulting current. The potential is swept between two set values at a constant scan rate, first in one direction (e.g., from a lower to a higher potential) and then back to the initial value [18]. When the applied potential reaches the energy level required to oxidize or reduce an analyte, a current peak is observed. The resulting plot of current versus potential is called a cyclic voltammogram [81].
For a reversible, diffusion-controlled redox reaction, the Randles-Ševčík equation (at 25 °C) describes the peak current (Ip):
Ip = (2.69 × 10^5) * n^(3/2) * A * D^(1/2) * C * υ^(1/2) [19]
where:
nis the number of electrons transferredAis the electrode area (cm²)Dis the diffusion coefficient (cm²/s)Cis the concentration (mol/mL)υis the scan rate (V/s)
The following diagram illustrates the workflow for a CV-based assessment, from experimental setup to data interpretation.
Diagram 1: CV Assessment Workflow for Flow Battery Molecules.
Quantitative Criteria for Electrochemical Reversibility
A reversible redox couple exhibits fast electron transfer kinetics at the electrode interface. The quantitative criteria for assessing reversibility are summarized in Table 1.
Table 1: Quantitative Criteria for Electrochemical Reversibility from Cyclic Voltammetry
| Parameter | Mathematical Expression | Criteria for Reversibility | Information Obtained |
|---|---|---|---|
| Peak Potential Separation | ΔEp = |Epa - Epc| | ΔEp ≈ 59/n mV (at 25°C) [81] | Electron transfer kinetics; ideal Nernstian behavior. |
| Peak Current Ratio | Ipa / Ipc | Ipa / Ipc ≈ 1 [19] | Chemical reversibility; stability of the oxidized/reduced form. |
| Peak Current vs. Scan Rate | Ip ∝ υ^(1/2) | Linear relationship of Ip vs. υ^(1/2) [19] | Diffusion-controlled process (as opposed to surface adsorption). |
| Peak Potential vs. Scan Rate | Epa and Epc | Independent of scan rate [18] | Fast electron transfer kinetics. |
| Half-Peak Potential | Epa - Ep/2 | Epa - Ep/2 > 56.5/n mV [81] | Number of electrons involved (n). |
For a reversible system, the formal reduction potential (E°') is the midpoint of the anodic and cathodic peak potentials: E°' = (Epa + Epc)/2 [81]. Deviations from these criteria indicate quasi-reversible or irreversible electron transfer, often associated with slow kinetics or coupled chemical reactions that consume the redox species.
Assessing Molecular Stability and Degradation
Beyond reversibility, the structural and chemical stability of redox-active molecules under operational conditions is critical for long-lasting flow batteries. Capacity fade in aqueous organic redox flow batteries (AORFBs) often stems from irreversible chemical reactions, such as the geminal diol formation in quinones or nucleophilic attacks on unsubstituted carbon sites [129] [130]. Cyclic voltammetry can diagnose these failure modes.
Diagnostic Protocols for Stability
- Multi-Cycle CV Analysis: Conducting consecutive CV cycles reveals molecular stability. A stable molecule will show overlapping voltammograms with minimal changes in peak current and potential. A gradual decrease in peak current indicates degradation of the active material [18].
- Scan Rate Dependence for Kinetic Analysis: Using the Randles-Ševčík equation, the diffusion coefficient (D) can be calculated from the slope of the Ip vs. υ^(1/2) plot. A significant change in D after cycling may indicate molecular aggregation or decomposition [19].
- Post-Mortem Analysis: Correlate CV data with ex-situ spectroscopic techniques. For instance, after observing capacity fade, use NMR or FT-IR to identify decomposition products like those from Michael addition or gem diol formation in benzoquinone derivatives [130].
Case Study: Hydroxylated Benzoquinones
Research on hydroxylated p-benzoquinone (p-BQ) derivatives demonstrates how CV informs stability. Introducing electron-donating hydroxyl (-OH) groups onto the BQ core improves electrochemical stability by increasing electron density and delocalizing charges [130]. However, over-substitution (e.g., tetrahydroxy-BQ) can cause electrostatic repulsion, destabilizing the molecule. CV shows that instability primarily arises from electrochemically irreversible redox reactions, not just the alkaline environment itself [130]. This highlights the need for an optimal functionalization strategy.
Experimental Protocol: CV for Flow Battery Molecules
Research Reagent Solutions
Table 2: Essential Materials and Reagents for CV Experiments
| Item | Specification / Example | Function / Purpose |
|---|---|---|
| Potentiostat | Instrument to control potential and measure current. | |
| Electrochemical Cell | 3-electrode configuration (Working, Reference, Counter) | Provides a controlled environment for the redox reaction. |
| Working Electrode | Glassy Carbon (for organic molecules), Platinum | Surface where the redox reaction of interest occurs. |
| Reference Electrode | Ag/AgCl (aqueous), SCE, Li/Li+ (non-aqueous) | Provides a stable, known reference potential. |
| Counter Electrode | Platinum wire or mesh | Completes the electrical circuit, allowing current to flow. |
| Redox-Active Species | e.g., Benzoquinone derivatives, Viologens [131] | The molecule under investigation. |
| Supporting Electrolyte | e.g., KCl, LiClO4, KOH (≥1 M) [26] | Provides ionic conductivity; minimizes resistive drop. |
| Solvent | Water, Acetonitrile, etc. | Dissolves the active species and supporting electrolyte. |
Step-by-Step Procedure
- Solution Preparation: Prepare a solution containing the redox-active molecule (typically 1-5 mM) in a suitable solvent with a supporting electrolyte (≥0.1 M) to ensure sufficient conductivity [26]. For aqueous organic flow battery studies, this might involve alkaline KOH solution for molecules like hydroxylated benzoquinones [130].
- Electrode Setup: Insert the working, reference, and counter electrodes into the cell. Prior to each experiment, polish the working electrode (e.g., glassy carbon) with alumina slurry (e.g., 0.05 µm) on a microcloth pad, then rinse thoroughly with deionized water and solvent [19].
- Purging: Sparge the solution with an inert gas (e.g., N2, Ar) for at least 10-15 minutes to remove dissolved oxygen, which can interfere with measurements.
- Instrument Calibration: Ensure the potentiostat is calibrated. Set the initial parameters: initial potential (Ei), switching potentials (E1, E2), and scan rate (υ). The potential window should be wide enough to fully observe the redox events of interest.
- Data Acquisition:
- Run an initial CV at a moderate scan rate (e.g., 100 mV/s) to identify the approximate redox potentials.
- To assess reversibility, run CVs at multiple scan rates (e.g., 10, 25, 50, 100, 200, 500 mV/s).
- To assess stability, run a series of consecutive CV cycles (e.g., 20-50 cycles) at a fixed scan rate.
- Data Analysis:
- For each voltammogram, record the anodic peak potential (Epa), cathodic peak potential (Epc), anodic peak current (Ipa), and cathodic peak current (Ipc).
- Calculate ΔEp and the Ipa/Ipc ratio for each scan rate.
- Plot Ip vs. υ^(1/2) to verify diffusion control.
- For multi-cycle data, plot normalized peak current versus cycle number to visualize degradation.
Advanced Application: Multi-Electron Transfer Systems
A promising route to higher energy density in flow batteries is using organic species capable of multi-electron transfer per molecule, such as viologens, quinones, and azines [131]. CV is indispensable for characterizing these complex systems. A single molecule may exhibit two or more distinct redox couples in its voltammogram. The reversibility criteria in Table 1 must be applied to each individual redox couple. For example, viologens show two sequential one-electron reductions (V²⁺ → V•⁺ → V⁰), where the first is often highly reversible, but the second can be poorly reversible, limiting practical capacity [131]. CV helps diagnose the kinetic limitations of each step and guides molecular engineering to improve the reversibility of all redox events.
Cyclic voltammetry is a powerful and accessible tool for the fundamental assessment of redox-active molecules for flow batteries. By systematically applying the protocols and diagnostic criteria outlined here—focusing on peak potential separation, current ratios, scan rate dependence, and multi-cycle stability—researchers can quantitatively evaluate electrochemical reversibility and identify degradation pathways. This enables the rational design of more stable, high-performance organic molecules, such as optimally functionalized quinones, accelerating the development of next-generation energy storage systems.
Conclusion
Cyclic Voltammetry stands as a powerful and versatile technique for redox reaction analysis, bridging fundamental thermodynamic study and practical application in biomedical research. Mastering its principles enables the determination of redox potentials and electron transfer kinetics, while a rigorous methodological approach ensures reliable data for applications ranging from drug quantification to neurotransmitter monitoring. Effective troubleshooting is paramount for data integrity, and validation through computational models and comparative analysis strengthens mechanistic insights. Future directions point toward the integration of nano-electrodes for single-molecule studies, advanced computational prediction of redox behavior for novel compounds, and the expanded use of FSCV and optical techniques for real-time, spatially resolved analysis in clinical and pharmaceutical settings, ultimately accelerating drug development and diagnostic innovation.
References
- 1. How to Choose Electrodes? A Practical Guide to the Three- ... [https://beyond-battery.com/en-us/blogs/beyond-battery-blogs/how-to-choose-electrodes-a-practical-guide-to-the-three-electrode-system]
- 2. Three Electrode System: The Key To Electrochemical ... [https://iestbattery.com/case/introduce-the-three-electrode-system/]
- 3. The Electrodes [https://sop4cv.com/chapters/TheElectrodes.html]
- 4. A Guide to the Application and Customization of Three- ... [https://www.neware.net/news/three-electrode-battery-testing-guide/230/112.html]
- 5. Electrochemical Aptasensor Developed Using Two- ... [https://www.mdpi.com/2079-6374/13/1/1]
- 6. Electrochemistry in a Two- or Three-Electrode Configuration to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10141359/]
- 7. Three-Electrode Setups [https://pineresearch.com/support-article/three-electrode-setups/]
- 8. 3D printed electro-responsive system with programmable ... [https://www.sciencedirect.com/science/article/pii/S2590049824000468]
- 9. Cyclic Voltammetry Basic Principles, Theory & Setup [https://www.ossila.com/pages/cyclic-voltammetry]
- 10. Cyclic Voltammetry in Pharma Analysis | PDF [https://www.scribd.com/document/466000558/An-overview-on-cyclic-voltammetry-and-its-application-in-pharmaceutical-analysis]
- 11. Cyclic Voltammetry - Data Analysis [https://www.basinc.com/manuals/EC_epsilon/Techniques/CycVolt/cv_analysis]
- 12. Generate Tafel Plots in AfterMath from CV or LSV Data [https://pineresearch.com/support-article/generate-tafel-plots-aftermath/]
- 13. Nernst Equation - Chemistry LibreTexts [https://chem.libretexts.org/Bookshelves/Analytical_Chemistry/Supplemental_Modules_(Analytical_Chemistry)/Electrochemistry/Nernst_Equation]
- 14. Nernst equation [https://en.wikipedia.org/wiki/Nernst_equation]
- 15. Nernst Equation - Chemistry Textbook [https://georgiasouthern.libguides.com/c.php?g=943952&p=6804656]
- 16. The Nernst Equation [https://sop4cv.com/chapters/NernstEquation.html]
- 17. Cyclic Voltammetry Part 1: Fundamentals [https://www.jstage.jst.go.jp/article/electrochemistry/90/10/90_22-66082/_html/-char/en]
- 18. Cyclic Voltammetry - CV Electrochemical Technique ... [https://www.gamry.com/electrochemistry-applications/cv-cyclic-voltammetry/]
- 19. Cyclic Voltammetry and Chronoamperometry: Mechanistic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10842901/]
- 20. Nernst Equation [https://chem.libretexts.org/Bookshelves/Analytical_Chemistry/Supplemental_Modules_(Analytical_Chemistry)/Electrochemistry/Basics_of_Electrochemistry/Electrochemistry/Nernst_Equation]
- 21. Randles–Sevcik equation [https://en.wikipedia.org/wiki/Randles%E2%80%93Sevcik_equation]
- 22. Randles–Sevcik equation [https://grokipedia.com/page/Randles%E2%80%93Sevcik_equation]
- 23. Understanding the Randles-Sevcik Equation [https://maciassensors.com/the-randles-sevcik-equation/]
- 24. Shedding light on the calculation of electrode electroactive ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10244294/]
- 25. Randles–Sevcik Equation Calculator [https://www.calctool.org/physical-chemistry/randles-sevcik-equation]
- 26. Cyclic voltammetry analysis of mercuric chloride redox ... [https://www.sciencedirect.com/science/article/abs/pii/S016773222402230X]
- 27. Randles–Ševčík Equation [https://pineresearch.com/support-article/randles-sevcik-equation/]
- 28. Comparative investigation of free radical scavenging and ... [https://www.nature.com/articles/s41598-025-12731-y]
- 29. Cyclic voltammetry for antioxidant assessment of moth ... [https://link.springer.com/article/10.1007/s11694-025-03266-x]
- 30. Optical Voltammetry of redox processes inside a nanohole ... [https://arxiv.org/html/2509.01815v1]
- 31. Electrochemical Reversibility [https://sop4cv.com/chapters/ElectrochemicalReversibility.html]
- 32. 2. Reversibility – Chemical vs. Electrochemical [https://chem.libretexts.org/Bookshelves/Analytical_Chemistry/Supplemental_Modules_(Analytical_Chemistry)/Analytical_Sciences_Digital_Library/Courseware/Analytical_Electrochemistry%3A_The_Basic_Concepts/03_Fundamentals_of_Electrochemistry/B%3A_The_Electrode_Process/02_Reversibility__Chemical_vs._Electrochemical]
- 33. Cv slides | PPTX [https://www.slideshare.net/slideshow/cv-slides-242371974/242371974]
- 34. Evaluating Electrochemical Reversibility [https://sop4cv.com/chapters/EvaluatingElectrochemicalReversibility.html]
- 35. Simulation-Based Approach to Determining Electron ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5660319/]
- 36. Physical interpretation of cyclic voltammetry for measuring ... [https://www.sciencedirect.com/science/article/abs/pii/S0013468612000023]
- 37. EDL structure and peculiarities of ferricyanide cyclic ... [https://www.sciencedirect.com/science/article/abs/pii/S1388248115001265]
- 38. Molecular-scale insights into the electrical double layer at ... [https://www.nature.com/articles/s41467-024-54631-1]
- 39. Cyclic Voltammetry (CV) [https://pineresearch.com/support-article/cyclic-voltammetry-cv/]
- 40. Optimization of Electrolytes with Redox Reagents to ... [https://www.mdpi.com/2079-6374/13/12/999]
- 41. Understanding electrochemical reactions using density ... [https://pubs.rsc.org/en/content/articlehtml/2025/cp/d5cp01464f]
- 42. Impact of electrolyte composition on the reactivity of a ... [https://pubs.rsc.org/en/content/articlelanding/2016/an/c6an00203j]
- 43. Practical considerations for using redox probes in ... [https://www.sciencedirect.com/science/article/pii/S0013468624016104]
- 44. How to Perform Cyclic Voltammetry on a Polymer using a ... [https://www.ossila.com/pages/how-to-perform-cyclic-voltammetry-on-a-polymer-using-a-potentiostat]
- 45. Performing Cyclic Voltammetry Measurements Using 2450- ... [https://www.tek.com/en/documents/application-note/performing-cyclic-voltammetry-measurements-using-2450-ec-or-2460-ec-electr]
- 46. Theoretical approach to the elliptic cyclic voltammetry of ... [https://www.sciencedirect.com/science/article/abs/pii/S1572665725003686]
- 47. Electrochemical and theoretical studies of the interaction ... [https://www.nature.com/articles/s41598-024-52609-z]
- 48. Development of a Cyclic Voltammetric Method for the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12474011/]
- 49. Probing Redox Responses and DNA Interactions in Drug ... [https://www.mdpi.com/2813-2998/4/2/20]
- 50. Measurements: Electrochemical Cyclic Voltammetry [https://www.nanoscience.com/techniques/electrochemistry/electrochemical-measurements-cyclic-voltammetry/]
- 51. - Wikipedia Cyclic voltammetry [https://en.wikipedia.org/wiki/Cyclic_voltammetry]
- 52. What Can Electrochemical Methods Offer in Determining ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8201389/]
- 53. Comprehensive review of the electrochemical detection ... [https://link.springer.com/article/10.1007/s43621-025-01384-6]
- 54. Electrochemical Sensor for the Evaluation of Doxorubicin ... [https://www.mdpi.com/2227-9040/12/4/69]
- 55. Recent Advances in Electrochemical Sensing of Anticancer ... [https://www.chemmethod.com/article_193533.html]
- 56. Electrochemical monitoring of the interaction of doxorubicin ... [https://www.sciencedirect.com/science/article/abs/pii/S1567539403000409]
- 57. Electrochemical behavior and square wave voltammetric ... [https://pubmed.ncbi.nlm.nih.gov/14969334/]
- 58. Voltammetric Sensor for Doxorubicin Determination Based ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10459114/]
- 59. Voltammetric DNA Sensor Based on Redox-Active Dyes for ... [https://link.springer.com/article/10.1134/S1061934822010075]
- 60. Voltammetric detection of anticancer drug Doxorubicin at ... [https://www.sciencedirect.com/science/article/pii/S2666351120300334]
- 61. Electrochemical Determination of Doxorubicin in the ... [https://www.mdpi.com/2079-6374/15/1/60]
- 62. Cellulose-based electrochemical recognition of ... [https://www.sciencedirect.com/science/article/pii/S2666893925002683]
- 63. Fundamentals of Fast-Scan Cyclic Voltammetry for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7028514/]
- 64. Fast-scan cyclic voltammetry [https://en.wikipedia.org/wiki/Fast-scan_cyclic_voltammetry]
- 65. Applying a Fast-Scan Cyclic Voltammetry to Explore ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9100021/]
- 66. Fast-scan cyclic voltammetry - PEMP lab [https://depts.washington.edu/pemplab/?page=fscv]
- 67. Recent Advances in Fast-Scan Cyclic Voltammetry - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7028521/]
- 68. A fast scan cyclic voltammetric digital circuit with precise ... [https://www.sciencedirect.com/science/article/abs/pii/S0003267021005705]
- 69. Real-Time Fast Scan Cyclic Voltammetry Detection and ... [https://www.frontiersin.org/journals/bioengineering-and-biotechnology/articles/10.3389/fbioe.2020.602216/full]
- 70. Recent advances in fast-scan cyclic voltammetry [https://pubs.rsc.org/zh-CN/content/articlelanding/2020/an/c9an01925a]
- 71. Fast-scan Cyclic Voltammetry for the Characterization of ... [https://www.sciencedirect.com/science/article/pii/S2001037014000567]
- 72. Effectiveness of Cyclic Voltammetry in Evaluation of the ... [https://www.mdpi.com/2304-8158/13/6/906]
- 73. A Simple Electrochemical Method for the Rapid Estimation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3876568/]
- 74. Cyclic voltammetry to evaluate the antioxidant potential in ... [https://www.sciencedirect.com/science/article/abs/pii/S0039914016309997]
- 75. Electrochemical Methodologies for Investigating the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9220340/]
- 76. FIA-ECD and Cyclic Voltammetry for antioxidant ... [https://www.sciencedirect.com/science/article/abs/pii/S0026265X25014870]
- 77. Antioxidant Determining Using Electrochemical Method [https://www.mdpi.com/2624-8549/5/3/131]
- 78. Effectiveness of Cyclic Voltammetry in Evaluation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10969822/]
- 79. Eco-Friendly Voltammetric Techniques for Assessing ... [https://www.mdpi.com/2673-6918/5/4/51]
- 80. “Electrochemical Index” as a screening method to ... [https://www.sciencedirect.com/science/article/abs/pii/S0003267005003028]
- 81. Cyclic Voltammetry [https://chem.libretexts.org/Bookshelves/Analytical_Chemistry/Supplemental_Modules_(Analytical_Chemistry)/Instrumentation_and_Analysis/Cyclic_Voltammetry]
- 82. Optical Voltammetry of redox processes inside a nanohole with ... [https://www.alphaxiv.org/overview/2509.01815v1]
- 83. Iontronic microscopy of a tungsten microelectrode [https://pmc.ncbi.nlm.nih.gov/articles/PMC10568260/]
- 84. Material Characterization Methods for Investigating Charge ... [https://pubs.rsc.org/en/content/articlelanding/2025/nr/d5nr00649j]
- 85. Understanding linear sweep voltammetry and cyclic ... [https://www.metrohm.com/en/discover/blog/2025/lsv-and-cv-explained.html]
- 86. ExperimentCyclicVoltammetry Documentation [https://www.emeraldcloudlab.com/helpfiles/experimentcyclicvoltammetry]
- 87. Troubleshooting Cyclic Voltammetry and Voltammograms [https://www.ossila.com/pages/cyclic-voltammetry-troubleshooting]
- 88. Trouble shooting cyclic voltammetry [https://www.zimmerpeacock.com/2025/06/28/trouble-shooting-cyclic-voltammetry/]
- 89. A Novel Method Based on Cyclic Voltammetry for Probing ... [https://www.sciencedirect.com/science/article/abs/pii/S2468023025018590]
- 90. A Comprehensive Guide to Cyclic Voltammetry(CV) [https://www.neware.net/news/a-comprehensive-guide-to-cyclic-voltammetry-cv/230/110.html]
- 91. What is noise, and how to avoid noise in electrochemistry? [https://www.palmsens.com/knowledgebase-article/what-is-noise-and-how-to-avoid-noise-in-electrochemistry-demo-included/]
- 92. Strategies for low detection limit measurements with cyclic ... [https://pubmed.ncbi.nlm.nih.gov/1789456/]
- 93. Cyclic Voltammetry (CV) [https://pineresearch.com/uncategorized/pine-aftermath-echem-cyclic-voltammetry/]
- 94. Critical thinking on baseline corrections for electrochemical ... [https://www.sciencedirect.com/science/article/pii/S0926337322002922]
- 95. Cyclic voltammetry [https://grokipedia.com/page/Cyclic_voltammetry]
- 96. Cyclic voltammetry insights into Ni/Al-carbonate ... [https://www.nature.com/articles/s41598-025-96933-4]
- 97. Using Cyclic Voltammetry to Detect Iron Impurities During ... [https://www.gamry.com/application-notes/physechem/using-cyclic-voltammetry-to-detect-iron/]
- 98. an open source approach described for cyclic voltammetry [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d4sc08620a]
- 99. EC_electrode_handbook/Section 6. Polishing method [https://www.als-japan.com/2022.html]
- 100. A comparative characterization analysis of electrode materials ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11439030/]
- 101. Frequency-domain analysis of voltammetric signals [https://www.sciencedirect.com/science/article/pii/S0263224125030659]
- 102. Cyclic Voltammetry Uses | How to Read a Voltammogram [https://www.ossila.com/pages/cyclic-voltammetry-applications]
- 103. Machine-learning-guided design of electroanalytical pulse ... [https://pubs.rsc.org/en/content/articlehtml/2025/dd/d5dd00005j]
- 104. Performance evaluation of fast Fourier-transform ... [https://www.sciencedirect.com/science/article/abs/pii/S0003267010014765]
- 105. DPPH Radical Scavenging Assay [https://www.mdpi.com/2227-9717/11/8/2248]
- 106. DPPH Measurements and Structure—Activity Relationship ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10967577/]
- 107. Review Application and Development of Electrochemical ... [https://www.sciencedirect.com/science/article/abs/pii/S100068182400064X]
- 108. DPPH Scavenging Assay Protocol- Detailed Procedure [https://acmeresearchlabs.in/2024/02/23/dpph-scavenging-assay-protocol-detailed-procedure/]
- 109. 3.5. DPPH Radical Scavenging Assay [https://bio-protocol.org/exchange/minidetail?id=8600043&type=30]
- 110. Spectroscopy and Spectrophotometry : Principles and... | IntechOpen [https://www.intechopen.com/chapters/79874]
- 111. Spectrophoto meter | PPT [https://www.slideshare.net/slideshow/spectrophoto-meter-31242060/31242060]
- 112. A rapid and simplified DPPH assay for analysis of ... [https://www.sciencedirect.com/science/article/pii/S0026265X24009135]
- 113. Understanding electrochemical reactions using density ... functional [https://pubs.rsc.org/en/content/articlelanding/2025/cp/d5cp01464f]
- 114. Application of Density Functional Theory to Molecular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11989887/]
- 115. Benchmarking OMol25-Trained Models on Experimental ... [https://rowansci.com/publications/omol25-redox]
- 116. Predicting Redox Potentials With Rowan - by Corin Wagen [https://rowansci.substack.com/p/predicting-redox-potentials-with]
- 117. DFT based structural modeling of chemotherapy drugs via ... [https://www.nature.com/articles/s41598-025-97982-5]
- 118. A Voltammetric Perspective of Multi-Electron and Proton ... [https://www.frontiersin.org/journals/chemistry/articles/10.3389/fchem.2021.672831/full]
- 119. Free Energies of Proton-Coupled Electron Transfer Reagents ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9175307/]
- 120. Principles and Case Studies of Cyclic Voltammetry Testing ... [https://www.neware.net/news/principles-and-case-studies-of-cyclic-voltammetry-testing-with-multiple-scan-rat/230/127.html]
- 121. Assessing Principal Component Regression Prediction of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3182154/]
- 122. Direct and two-stage data analysis procedures based on ... [https://www.sciencedirect.com/science/article/abs/pii/S0039914005001499]
- 123. Relationships between PCA and PLS-regression [https://www.sciencedirect.com/science/article/abs/pii/S0169743913002189]
- 124. Electrochemical Fingerprint Analyses Using Voltammetry ... [https://www.mdpi.com/1420-3049/30/1/48]
- 125. Electroanalysis Advances in Pharmaceutical Sciences [https://www.mdpi.com/2673-4532/6/2/12]
- 126. 25.4: Cyclic Voltammetry [https://chem.libretexts.org/Bookshelves/Analytical_Chemistry/Instrumental_Analysis_(LibreTexts)/25%3A_Voltammetry/25.04%3A_Cyclic_Voltammetry]
- 127. What is CV? A comprehensive guide to Cyclic Voltammetry [https://www.biologic.net/topics/what-is-cv-a-comprehensive-guide-to-cyclic-voltammetry/]
- 128. Target analyte assisted sensitive electrochemical detection of ... [https://pubs.rsc.org/en/content/articlehtml/2025/lf/d5lf00006h]
- 129. Monitoring chemical processes in redox flow batteries ... [https://pubs.rsc.org/en/content/articlehtml/2025/ee/d5ee01311a]
- 130. Unveiling dominant impact of electrochemical stability on ... [https://www.sciencedirect.com/science/article/abs/pii/S0378775324007183]
- 131. Multi-redox Organic Species for High-Energy-Density ... [https://www.sciencedirect.com/science/article/abs/pii/S2405829725006828]