Beyond the Background: Advanced Strategies for Correcting Non-Faradaic Currents in Biomedical Electroanalysis
This article provides a comprehensive guide for researchers and drug development professionals on managing non-Faradaic (capacitive) currents, a major source of interference in electrochemical biosensing.
Beyond the Background: Advanced Strategies for Correcting Non-Faradaic Currents in Biomedical Electroanalysis
Abstract
This article provides a comprehensive guide for researchers and drug development professionals on managing non-Faradaic (capacitive) currents, a major source of interference in electrochemical biosensing. Covering foundational principles to cutting-edge applications, we explore the critical distinction between Faradaic and non-Faradaic processes and their impact on assay sensitivity in complex matrices like serum and blood. The content details innovative hardware and methodological solutions, including differential potentiostats and specialized measurement techniques, for real-time background suppression. It further offers practical troubleshooting and optimization strategies for electrode design and system validation, culminating in a comparative analysis of techniques to enhance the accuracy, reliability, and translational potential of electrochemical diagnostics and bioanalytical assays.
Understanding the Signal and the Noise: A Primer on Non-Faradaic Currents in Bioelectrochemistry
Fundamental Definitions
What is the core difference between a Faradaic and a non-Faradaic process?
A Faradaic process involves the transfer of charge (electrons) across the electrode-electrolyte interface, leading to oxidation or reduction reactions. In contrast, a non-Faradaic process involves the storage of charge at the interface without any electron transfer or change in the oxidation state of species [1] [2].
- Faradaic Process (Charge Transfer): This is an electron transfer reaction where ions in the electrolyte gain or lose electrons at the electrode surface. Examples include ions interacting directly with the electrode or molecules undergoing redox reactions. This process produces a faradaic current that is easily identified in techniques like cyclic voltammetry as distinct peaks [1].
- Non-Faradaic Process (Charge Storage): This process does not involve electron transfer reactions. Instead, charge is stored electrostatically through mechanisms like ion adsorption onto the electrode surface or intercalation into materials without changing their oxidation state. This generates a capacitive current, characterized by a rectangular shape in cyclic voltammetry, indicating simple charging and discharging [1] [2].
The table below summarizes the key characteristics:
Table 1: Core Characteristics of Faradaic and Non-Faradaic Processes
| Feature | Faradaic Process | Non-Faradaic Process |
|---|---|---|
| Charge Transfer | Yes, across the electrode interface | No, charge is stored at the interface |
| Redox Reactions | Occurs; oxidation states change | Does not occur; no change in oxidation state |
| Current Type | Faradaic current | Capacitive (non-faradaic) current |
| Cyclic Voltammetry Signature | Peaks representing redox reactions | Rectangular shape representing charging/discharging |
| Process Reversibility | Often chemically irreversible | Highly electrically reversible |
| Primary Mechanism | Electron transfer (oxidation/reduction) | Electrostatic attraction, ion adsorption, double-layer formation |
Troubleshooting Guides & FAQs
FAQ: Identifying Process Type
How can I tell if my measurement is dominated by a Faradaic or non-Faradaic signal?
You can distinguish them by analyzing your cyclic voltammetry (CV) data [1]:
- Faradaic Signal: Look for distinct, sharp peaks in the voltammogram. The position of these peaks corresponds to the redox potential of the electroactive species.
- Non-Faradaic Signal: Look for a rectangular, box-like shape in the voltammogram. This indicates a continuous charging and discharging of the electrical double-layer.
It is crucial to note that broad peaks in a CV do not automatically confirm a Faradaic process, as some capacitive materials can also exhibit such features [2].
Why is the capacitive (non-faradaic) current considered a key limitation in sensitive electrochemical assays?
The non-faradaic current acts as a significant, non-zero background signal. This is particularly problematic in modern assay systems, such as those using DNA monolayers, because it can [3]:
- Overwhelm the smaller faradaic current from the analyte of interest.
- Limit the signal-to-noise ratio, reducing assay sensitivity.
- Saturate the instrument's amplifiers, especially when using larger electrode surfaces to amplify the faradaic signal.
FAQ: Mitigating Non-Faradaic Current
What experimental strategies can I use to suppress non-faradaic current interference?
Several methodological and hardware approaches can be employed:
- Hardware Subtraction: Using a differential potentiostat (DiffStat) with two working electrodes. One electrode (W1) provides the analytical signal (faradaic + non-faradaic), while the other (W2) provides only the background (non-faradaic). The DiffStat subtracts the background from the analytical signal in real-time, outputting a signal predominantly composed of faradaic current [3].
- Pulsed Voltammetry Techniques: Using techniques like square-wave voltammetry (SWV) or differential pulse voltammetry, which can digitally subtract background currents during data processing [3].
- Control Electrode Surface Area: Since non-faradaic current is directly proportional to electrode surface area, reducing the area can lower the background. However, this also reduces the faradaic current, requiring more sensitive instrumentation [3].
My droplet-based electrochemical measurements (e.g., SECCM) show abnormally high activity at grain boundaries. Is this a Faradaic enhancement?
Not necessarily. Studies on polycrystalline platinum have shown that surface tension effects at grain boundaries can have a large conflating effect in droplet-based techniques like Scanning Electrochemical Cell Microscopy (SECCM), often producing false positives. The local curvature and wettability of the surface can alter the meniscus contact and the measured current, which may be mistaken for enhanced Faradaic (electrocatalytic) activity. Rigorous surface preparation and surface area correction techniques are required to confirm true intrinsic activity [4].
Experimental Protocols
Protocol 1: Differentiating Processes via Cyclic Voltammetry
This protocol provides a methodology to characterize an electrode material and identify the nature of its electrochemical response.
- 1. Objective: To distinguish between Faradaic and non-Faradaic processes using Cyclic Voltammetry (CV).
- 2. Materials:
- Potentiostat/Galvanostat
- Standard three-electrode cell
- Working Electrode (e.g., glassy carbon, gold, or material under study)
- Counter Electrode (e.g., platinum wire)
- Reference Electrode (e.g., Ag/AgCl)
- Electrolyte solution (e.g., 0.5 M H₂SO₄ or other suitable supporting electrolyte)
- 3. Methodology:
- Prepare the electrode surface according to standard procedures (e.g., polishing for solid electrodes).
- Set up the electrochemical cell with the working, counter, and reference electrodes immersed in the electrolyte.
- Program the potentiostat with a CV method. A typical initial scan would be from -0.2 V to +0.6 V vs. Ag/AgCl and back, at a scan rate of 50 mV/s.
- Run the CV and record the current response.
- Analyze the resulting voltammogram for the presence of peaks (indicative of Faradaic processes) or a rectangular shape (indicative of non-Faradaic, capacitive behavior) [1] [2].
- 4. Data Interpretation:
- Identify and note the number of peaks, their potential positions (Epa for anodic, Epc for cathodic), and their peak currents (ip).
- A large, rectangular current with no distinct peaks suggests the material is primarily capacitive.
Protocol 2: Hardware Suppression of Non-Faradaic Current
This protocol outlines the use of a differential potentiostat to minimize capacitive background in sensitive measurements, such as DNA-based hybridization assays [3].
- 1. Objective: To suppress non-faradaic current in real-time using a differential potentiostat (DiffStat) configuration.
- 2. Materials:
- Differential Potentiostat (DiffStat)
- Two electrochemical cells, each with a separate Working Electrode (W1 and W2)
- Split Reference Electrode and Counter Electrode to connect both cells
- For DNA sensing: Gold working electrodes functionalized with thiolated DNA monolayers.
- 3. Methodology:
- Cell Preparation: Fabricate W1 and W2 in two separate cells to prevent cross-contamination. The analyte (e.g., MB-DNA) is added only to W1. A carefully matched control (e.g., CTR-DNA) is added to W2 to ensure the non-faradaic components are identical [3].
- Instrument Setup: Connect W1 and W2 to the DiffStat's two working electrode inputs. The DiffStat utilizes matching transimpedance amplifier circuits for each electrode, with the outputs fed into a differential instrumentation amplifier for real-time analog subtraction [3].
- Measurement: Perform the electrochemical measurement (e.g., Chronoamperometry, CV, or SWV). The DiffStat will output a signal where the background capacitive current from W2 has been subtracted from the combined signal from W1.
- 4. Data Interpretation: Compare the output from the DiffStat with data from a conventional potentiostat. A successful implementation will show a significant reduction in the baseline current while preserving the faradaic peak current.
Visual Explanations
Diagram 1: Process Comparison and Measurement Setup
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 2: Key Materials and Their Functions in Electrochemical Research
| Item | Function / Application |
|---|---|
| Bismuth Electrode | An environmentally friendly alternative to mercury electrodes for sensitive tensiometric and electrochemical measurements [5]. |
| Quasi-Reference Counter Electrode (QRCE) | Integrated into nanopipette probes for techniques like SECCM, providing a compact reference and counter electrode system [4]. |
| Nanopipette Probe | A borosilicate glass pipette with a nanoscale tip (e.g., 200 nm) used in SECCM to isolate and measure tiny areas of an electrocatalyst surface [4]. |
| Methylene Blue (MB) | A common redox reporter molecule used in DNA-based electrochemical assays (e.g., E-DNA biosensors) to generate a faradaic current [3]. |
| Thiolated DNA | Used to form self-assembled monolayers (SAMs) on gold electrodes, which serve as the foundation for many modern bioanalytical sensors [3]. |
| Differential Potentiostat (DiffStat) | Specialized hardware that uses two working electrodes to perform real-time analog subtraction of non-faradaic background current [3]. |
Theoretical Foundations: The Electrical Double Layer
What is the electrical double layer and how does it form?
The electrical double layer (EDL) is a structure that forms at the interface between an electrode and an electrolyte solution when the two are brought into contact [6]. This interface is fundamental to all electrochemical systems. When a charged electrode is introduced into an electrolyte, the electric field generated by the electrode surface acts on the charged ions in the solution, causing them to rearrange [7]. This results in a structured region of ions that screens the electrode's charge from the bulk solution.
The formation follows this sequence:
- The electrode possesses a surface charge (either positive or negative).
- Ions of opposite charge (counter-ions) in the solution are electrostatically attracted to the electrode surface.
- Ions of the same charge (co-ions) are repelled from the surface.
- This rearrangement creates a "double layer" of charge: one layer on the electrode surface and a second layer of ions in the solution.
What are the key models describing the EDL structure?
Our understanding of the EDL has evolved through several key historical models, summarized in the table below.
Table 1: Historical Evolution of Electrical Double Layer Models
| Model | Key Proposer(s) | Year(s) | Core Concept | Limitations |
|---|---|---|---|---|
| Helmholtz | Hermann von Helmholtz | 1853 | A rigid, molecular capacitor with two layers of opposite charge [6] [8]. | Does not consider thermal motion of ions; only valid for high electrolyte concentrations [6]. |
| Gouy-Chapman | Gouy & Chapman | 1910, 1913 | A diffuse layer where ion distribution is governed by electrostatic forces and thermal motion [6] [8]. | Predicts impossibly high ion densities near the electrode for high potentials [6]. |
| Stern | Otto Stern | 1924 | A hybrid model: a rigid Stern layer of adsorbed ions and a diffuse Gouy-Chapman layer [6] [8]. | Treats ions as point charges; assumes constant permittivity [6]. |
| Grahame | D.C. Grahame | 1947 | Divided the Stern layer into the Inner Helmholtz Plane (IHP) (specifically adsorbed, desolvated ions) and the Outer Helmholtz Plane (OHP) (solvated ions at their closest approach) [6]. | - |
| Bockris/Devanathan/Müller (BDM) | Bockris, Devanathan, & Müller | 1963 | Incorporated the specific orientation of solvent molecules (e.g., water) at the electrode surface, which influences the interface's permittivity [6]. | - |
The following diagram illustrates the structure of the electrical double layer, integrating concepts from the Stern, Grahame, and BDM models.
Diagram 1: Structure of the Electrical Double Layer and Potential Decay
What is the direct link between the EDL and non-Faradaic current?
The non-Faradaic current (also called charging or capacitive current) originates directly from the capacitor-like nature of the electrical double layer [9] [10].
- The Double Layer as a Capacitor: The electrode-electrolyte interface behaves like a capacitor, often termed the double-layer capacitance (Cdl). The charged electrode and the layer of counter-ions in the solution act as the two "plates" of this capacitor, separated by a molecular-scale distance [9] [8].
- Charging Current: When the potential applied to the electrode is changed, the charge stored on this "capacitor" must also change. The current that flows to effect this change in charge—by rearranging ions at the interface—is the non-Faradaic current [10]. It is described by the equation:
i_c = C_dl * (dE/dt)wherei_cis the capacitive current,C_dlis the double-layer capacitance, anddE/dtis the rate of change of the applied potential [9] [11]. - Key Characteristic: In a purely non-Faradaic process, no electrons transfer across the electrode-electrolyte interface to cause oxidation or reduction of solution species. The charge transfer is purely electrostatic [9] [2].
Table 2: Contrasting Faradaic and Non-Faradaic Processes
| Feature | Faradaic Process | Non-Faradaic (Capacitive) Process |
|---|---|---|
| Definition | Electron transfer across the interface causes oxidation/reduction reactions [9]. | Electrostatic charging/discharging of the double-layer capacitor; no redox chemistry [9] [2]. |
| Governed by | Faraday's Law (amount of chemical change ∝ current) [9]. | Capacitance law (q = C×E) [10]. |
| Current Type | Faradaic current. | Charging (capacitive) current. |
| Effect on Solution | Composition changes (Ox Red). | No net change in solution composition; only ion rearrangement at the interface. |
| Persistence | Continuous current at constant potential (if mass transport is sustained). | Transient current only when potential is changing (dE/dt ≠ 0) [11]. |
Troubleshooting Guide: Common Experimental Issues
How can I minimize the interference of non-Faradaic current in my measurements?
Non-Faradaic current is a major source of background interference, limiting the sensitivity and detection limit of electrochemical assays, particularly those using DNA monolayers or other surface-bound systems [3]. The table below outlines common problems and solutions.
Table 3: Troubleshooting Non-Faradaic Current Interference
| Problem | Possible Cause | Solutions & Recommendations |
|---|---|---|
| High background in cyclic voltammetry (CV) or square-wave voltammetry (SWV) | Large C_dl and fast scan rates (dE/dt) leading to large i_c [3] [10]. |
1. Use pulsed techniques (e.g., SWV, differential pulse voltammetry) which discriminate against capacitive current [3]. 2. Reduce scan rate to lower i_c (but this also lowers faradaic signal). 3. Employ background subtraction in software or, ideally, via hardware (see Section 2.2). |
| Low signal-to-noise (S/N) ratio in chronoamperometry | Non-faradaic decay current obscures the faradaic response [3]. | 1. Use a longer delay time before current measurement to allow capacitive decay. 2. Apply data fitting to model and subtract the decaying background [3]. |
| Instrument amplifier saturation | Using a large electrode surface area, which increases both faradaic and non-faradaic currents [3]. | 1. Reduce working electrode area (but this also reduces desired faradaic signal). 2. Implement hardware current subtraction (e.g., a differential potentiostat) to remove the capacitive component before amplification [3]. |
| Difficulty detecting low analyte concentrations | Faradaic signal is small and obscured by the capacitive background [3]. | 1. Optimize electrochemical technique parameters (pulse height, step potential, frequency). 2. Lower the measurement's time constant to better resolve faradaic and non-faradaic components. 3. Use a differential potentiostat (DiffStat) for real-time analog suppression of capacitive current [3]. |
What advanced hardware solutions exist for non-Faradaic current suppression?
A powerful approach is the use of a differential potentiostat (DiffStat), which suppresses non-faradaic current through real-time analog subtraction [3].
- Concept: The DiffStat uses two working electrodes (W1 and W2) in a single electrochemical cell.
- W1 is the functional sensor (e.g., with a DNA monolayer and redox reporter).
- W2 is a nearly identical "blank" electrode (e.g., with a DNA monolayer but no redox reporter).
- How It Works: Since both electrodes have the same double-layer structure, they experience nearly identical non-faradaic currents. The DiffStat circuitry measures the current at both electrodes simultaneously and subtracts the background current (from W2) from the total current (from W1) in real-time [3]. The output is a signal predominantly composed of faradaic current.
- Benefits:
- Improved S/N Ratio: Can suppress capacitive background by ~5-fold or more [3].
- Larger Accessible Electrodes: Prevents amplifier saturation, allowing the use of larger electrodes for bigger faradaic signals.
- Simplified Data Processing: Outputs a "cleaner" signal, reducing the need for complex digital background fitting [3].
The workflow and configuration of a DiffStat are illustrated below.
Diagram 2: Comparison of Conventional and Differential Potentiostat Configurations
Experimental Protocols & Methodologies
How do I characterize the double-layer capacitance (Cdl) of my electrode material?
Objective: To determine the double-layer capacitance, a key parameter defining the magnitude of non-faradaic current, for a modified or unmodified electrode.
Principle: In a potential window where no faradaic reactions occur, the electrode-electrolyte interface behaves like a pure capacitor. By measuring the current response to different potential scan rates in this "non-faradaic" region, Cdl can be calculated.
Materials & Equipment:
- Potentiostat (standard three-electrode system)
- Working Electrode (material under study)
- Counter Electrode (e.g., Pt wire)
- Reference Electrode (e.g., Ag/AgCl)
- Electrolyte solution (e.g., 0.1 M KCl or other supporting electrolyte without redox-active species)
Procedure:
- Setup: Place the working electrode in the electrolyte solution and connect the three-electrode system to the potentiostat.
- Select a Non-Faradaic Potential Window: Using cyclic voltammetry (CV), identify a potential range (e.g., ±50 mV around the open circuit potential) where no redox peaks are observed. This confirms the absence of faradaic processes.
- Record CVs at Multiple Scan Rates: In this selected potential window, record CV scans at several different scan rates (e.g., 10, 25, 50, 75, 100 mV/s).
- Measure Charging Current: For each scan rate, measure the current (
i_c) at a fixed potential within the window (e.g., at the middle of the potential range). - Plot and Calculate: Plot the absolute value of the charging current (
|i_c|) against the scan rate (v). The relationship should be linear:i_c = C_dl * v. The double-layer capacitance (Cdl) is the slope of this line.
Notes:
- For a flat electrode, Cdl is often reported in
F/cm². Ensure you know the precise geometric area of your electrode. - For porous or high-surface-area materials, the measured value is the total capacitance, which can be used to estimate the electroactive surface area (ECSA).
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Materials for Investigating and Managing Non-Faradaic Effects
| Reagent/Material | Function/Description | Example Use Case |
|---|---|---|
| Supporting Electrolyte (e.g., KCl, NaNO₃, KPF₆) | Carries current in solution without participating in faradaic reactions. High concentration minimizes solution resistance and defines the Debye length (double-layer thickness) [7]. | Used in all electrochemical experiments to control ionic strength and minimize IR drop. |
| Redox-Inactive Probe Molecules | Molecules that do not undergo electron transfer in the studied potential window. Used to characterize the capacitive properties of the interface without faradaic interference. | Used in the CV scan-rate method to determine Cdl (Protocol 3.1). |
| Self-Assembled Monolayer (SAM) Forming Thiols (e.g., 6-mercapto-1-hexanol) | Forms an organized, dense monolayer on gold electrodes. This passivates the surface and drastically reduces Cdl by moving the diffuse layer further from the electrode [3]. | Used in E-DNA and E-AB biosensors to minimize non-faradaic background and prevent nonspecific adsorption [3]. |
| Differential Potentiostat (DiffStat) | Specialized hardware that uses two working electrodes for real-time analog subtraction of capacitive current [3]. | For high-sensitivity measurements in complex matrices (e.g., serum, whole blood) where background currents are high and unstable [3]. |
| Redox Reporters with Large ∆Ep (e.g., Methylene Blue) | Molecules with a significant potential difference between oxidation and reduction peaks. This allows analytical techniques (like SWV) to be performed at potentials away from the large current swings of the redox event, reducing overlap with capacitive currents [3]. | Used as labels in DNA-based electrochemical sensors to generate a measurable faradaic signal [3]. |
Why Non-Faradaic Currents Limit Sensitivity and Accuracy in Biomedical Sensing
FAQ: What are Non-Faradaic Currents and Why are They Problematic?
Q1: What is the fundamental difference between a Faradaic and a non-Faradaic process? A Faradaic process involves the actual transfer of charged particles (electrons) across the electrode-electrolyte interface, leading to a reduction-oxidation (redox) reaction. After applying a constant current, the electrode charge, voltage, and composition reach constant values [2]. In contrast, a non-Faradaic process (also called capacitive) does not involve charge transfer across the interface; instead, charge is progressively stored at the electrode surface, much like a capacitor charging and discharging [12] [2]. This charging of the electrical double layer at the interface is the source of non-Faradaic, or capacitive, current [3].
Q2: How exactly do non-Faradaic currents interfere with sensor measurements? Non-Faradaic currents act as a significant, non-zero baseline interference that obscures the desired analytical signal—the Faradaic current [3]. This interference has several direct consequences:
- Reduced Signal-to-Background Ratio: The capacitive current can overwhelm the smaller Faradaic current generated by the target biomarker, making the signal difficult to distinguish from noise [3].
- Limited Electrode Surface Area: The magnitude of the non-faradaic current is directly proportional to the electrode surface area. Using larger electrodes to amplify the Faradaic signal simultaneously amplifies the background capacitive current, which can saturate the instrument's amplifiers and limit detection sensitivity [3].
- Narrowed Detection Range: The large background can compress the dynamic range of the sensor, limiting its ability to detect a wide concentration of analytes [3].
TROUBLESHOOTING GUIDE: Mitigating Non-Faradaic Currents
Problem: My electrochemical biosensor has a high background signal, limiting its detection sensitivity. Solution: Here are proven strategies to suppress non-Faradaic interference:
- Solution 1: Implement Hardware Subtraction. Utilize a differential potentiostat (DiffStat). This instrument uses two working electrodes: one experimental sensor (W1) and one "blank" background electrode (W2). The circuitry performs real-time, analog subtraction of the capacitive current from W2 from the total current from W1, outputting a signal that is predominantly the desired Faradaic current. This method has been shown to suppress capacitance current by approximately 5-fold in chronoamperometry measurements [3].
- Solution 2: Optimize Electrode Geometry and Material. For capacitive (non-Faradaic) biosensors, the design of interdigitated electrodes (IDEs) is critical. Research shows that reducing the gap between the fingers of the IDE significantly enhances sensitivity. One study found that a 3 μm gap configuration could detect a target concentration of 50 ng/mL, a threshold unattainable by designs with 4 μm or 5 μm gaps [13]. Furthermore, be aware that electrode material stability impacts performance. Aluminum electrodes, for instance, are susceptible to surface corrosion in extreme pH environments, which degrades their performance in both Faradaic and non-Faradaic measurements [14].
- Solution 3: Leverage Underutilized Impedimetric Parameters. When using a non-Faradaic EIS biosensor, do not rely solely on measuring capacitance. A study on interleukin-8 (IL-8) detection demonstrated that the imaginary impedance (Zimag) parameter provided the best performance, with a limit of detection of 90 pg/mL and the highest sensitivity at 13.1 kΩ/log(ng/mL), outperforming the more commonly measured capacitance [15].
Experimental Protocols for Advanced Research
Protocol 1: Differentiating Faradaic and Non-Faradaic Processes via Cyclic Voltammetry (CV) This protocol helps characterize the nature of your electrode process.
- 1. Objective: To determine whether an electrode material exhibits primarily Faradaic or non-Faradaic behavior.
- 2. Materials:
- Potentiostat
- Standard three-electrode setup (Working, Counter, Reference electrodes)
- Electrode material under test
- Electrolyte solution (e.g., PBS or with a redox couple like [Fe(CN)₆]³⁻/⁴⁻)
- 3. Methodology:
- Prepare the electrode and place it in the electrolyte solution.
- Run a cyclic voltammetry scan at a moderate scan rate (e.g., 50 mV/s) over a suitable potential window.
- Observe the shape of the CV curve.
- 4. Data Interpretation:
- Faradaic Dominated: Look for distinct, sharp oxidation and reduction peaks corresponding to redox reactions [2].
- Non-Faradaic Dominated: The CV diagram will have a more rectangular, box-like shape, characteristic of capacitive charging and discharging. Note that broad peaks can sometimes appear in capacitive materials, so shape alone is not definitive; the underlying charge storage mechanism is key [2].
Protocol 2: Detecting a Biomarker using a Non-Faradaic Impedimetric Biosensor This protocol outlines the steps for a label-free capacitive biosensor, optimized for an interdigitated electrode (IDE).
- 1. Objective: To functionalize an IDE and detect a specific biomarker (e.g., IL-8) by monitoring changes in non-Faradaic impedance parameters [15].
- 2. Research Reagent Solutions:
| Reagent | Function in the Experiment |
|---|---|
| Gold Interdigitated Electrodes (Au-IDEs) | The transducer platform; its surface is modified to capture the target. |
| Cysteamine | Forms a self-assembled monolayer (SAM) on the gold surface, providing terminal amine groups for further cross-linking [15]. |
| Glutaraldehyde | A crosslinker that reacts with the amine groups from cysteamine, providing aldehyde groups for antibody immobilization [15]. |
| IL-8 Antibodies | The biorecognition element that selectively binds to the IL-8 antigen target [15]. |
| Bovine Serum Albumin (BSA) | Used to block non-specific binding sites on the electrode surface, reducing false-positive signals [15]. |
| Phosphate-Buffered Saline (PBS) | Provides a stable ionic environment for electrochemical measurements [15]. |
- 3. Methodology:
- Biofunctionalization:
- SAM Formation: Incubate the clean Au-IDE in a 1 mM ethanolic cysteamine solution for 1 hour. Rinse with ethanol and dry with N₂ [15].
- Cross-linking: Apply 50 μL of 2.5% glutaraldehyde in PBS to the surface. Incubate for 1 hour. Rinse with DI water and dry with N₂ [15].
- Antibody Immobilization: Apply 50 μL of a 1 μg/mL IL-8 antibody solution. Incubate for 1 hour. Rinse with DI water [15].
- Surface Blocking: Apply 50 μL of 5% BSA in PBS. Incubate for 30 minutes. Rinse with DI water and dry with N₂ [15].
- Non-Faradaic EIS Measurement:
- Baseline: Place 50 μL of PBS on the functionalized IDE. Measure the open-circuit potential (OCP) for 100 seconds to ensure equilibrium [15].
- Acquisition: Conduct EIS measurements with a zero DC potential (relative to OCP) to operate in a non-Faradaic mode. A small AC voltage (e.g., 10 mV) is applied across a frequency range (e.g., 100 Hz to 1 MHz) [15].
- Detection: Replace PBS with the sample solution containing the IL-8 antigen. Repeat the EIS measurement.
- Biofunctionalization:
- 4. Data Analysis:
- Extract key impedance parameters from the spectra: Imaginary Impedance (Zimag), Impedance Magnitude (Zmod), Capacitance, and Real Impedance (Zreal).
- Construct a calibration curve by plotting the change in the parameter (e.g., Zimag) against the logarithm of the antigen concentration. As per recent findings, Zimag is likely to provide the highest sensitivity and lowest limit of detection [15].
Performance Data of Mitigation Strategies
The table below summarizes quantitative data from recent studies on improving sensor performance by addressing non-Faradaic currents.
Table 1: Quantitative Performance of Different Strategies for Managing Non-Faradaic Effects
| Strategy | Experimental Context | Key Performance Metric | Result | Source |
|---|---|---|---|---|
| Hardware Subtraction (DiffStat) | DNA monolayer-based sensor using Chronoamperometry (CA) | Capacitive Current Suppression | ~5-fold reduction | [3] |
| Electrode Geometry Optimization | IDE biosensor for antibody detection | Limit of Detection (LoD) | 3 μm gap: 50 ng/mL (Lowest achievable) | [13] |
| Parameter Selection (Non-Faradaic EIS) | Au-IDE biosensor for IL-8 detection | Limit of Detection (LoD) | Zimag: 90 pg/mL; Capacitance: 140 pg/mL | [15] |
| Parameter Selection (Non-Faradaic EIS) | Au-IDE biosensor for IL-8 detection | Sensitivity | Zimag: 13.1 kΩ/log(ng/mL) (Highest) | [15] |
Technological Workflows
The following diagrams illustrate the core concepts and experimental workflows discussed in this guide.
Diagram 1: Fundamental processes and the problem of non-Faradaic current.
Diagram 2: DiffStat workflow for hardware-level current suppression.
Troubleshooting Guide: Improving Signal-to-Background Ratios
Problem: High non-faradaic (capacitive) background currents are obscuring my analytical signal.
Non-faradaic or capacitive current originates from the formation of a double layer at the electrode and monolayer surface, creating a time-dependent background that interferes with the analytical faradaic current. This is a key factor limiting sensitivity in many electrochemical assays, especially those based on DNA monolayers [3].
Solutions:
- Implement a Differential Potentiostat (DiffStat): This hardware solution uses two working electrodes (W1 for signal, W2 for background) with matching transimpedance amplifier circuits that feed into a differential instrumentation amplifier. The signals from both electrodes are collected simultaneously and analog-subtracted, effectively suppressing the capacitive current at the source [3]. This method has been shown to suppress baseline capacitance current by approximately 5-fold in chronoamperometry and can make larger electrodes and higher sensitivity settings accessible [3].
- Optimize Electrochemical Techniques: Use pulse voltammetry techniques like Square-Wave Voltammetry (SWV), which are designed to discriminate against capacitive currents through digital subtraction during data processing [3].
- Control Electrode Surface Area: Since non-faradaic current is directly proportional to electrode surface area, consider using smaller electrodes. Be aware that this will also reduce your faradaic current, requiring higher sensitivity instrumentation [3].
- Refine Data Processing: Apply digital background subtraction or fitting algorithms to chronoamperometric data during analysis to remove the non-faradaic component [3].
Problem: My signal is lost in environmental and instrumental noise.
Environmental electrical interference is a pervasive source of artifacts, often manifesting as 50/60 Hz mains hum or high-frequency hash.
Solutions:
- Use Differential Amplification: Employ a differential amplifier with a high Common-Mode Rejection Ratio (CMRR > 100 dB). This setup measures the voltage difference between an active electrode and a reference electrode, subtracting noise common to both inputs (like line noise) [16].
- Establish Proper Grounding and Shielding:
- Single-Point Grounding: Connect all components to a single common earth ground point to prevent ground loops, a major source of 60 Hz hum [16].
- Faraday Cage: Use a conductive enclosure around the experimental preparation and headstage to attenuate high-frequency electromagnetic interference (EMI) and radio frequency interference (RFI) [16].
- Cable Management: Use shielded, twisted-pair cables and avoid running power and signal lines in parallel to minimize inductive and capacitive coupling [16].
- Apply Digital Filtering (Post-Acquisition):
- Notch Filter: Precisely remove 50/60 Hz mains interference. Use cautiously as it can introduce artifacts [16].
- Low-Pass Filter: Attenuate high-frequency noise (e.g., thermal noise). Set the cutoff just above the fastest frequency component of your biological signal [16].
- High-Pass Filter: Remove slow baseline drift caused by electrode instability or temperature changes [16].
- Use Signal Averaging: For signals time-locked to a stimulus (e.g., evoked potentials), repeatedly record and average the responses. Uncorrelated random noise averages toward zero, while the time-locked signal is enhanced, improving the signal-to-noise ratio (SNR) proportionally to the square root of the number of trials [17] [16].
Frequently Asked Questions (FAQs)
Q1: How does electrode surface area affect my signal and background, and what are the trade-offs?
The electrode surface area has a direct and often competing impact on both faradaic (signal) and non-faradaic (background) currents.
- Faradaic Current: The analytical signal from your redox reaction is generally proportional to the electrode surface area. A larger area provides more reaction sites, increasing your signal [18].
- Non-Faradaic (Capacitive) Current: This background current is also directly proportional to the electrode surface area. A larger electrode creates a larger double-layer capacitance, leading to a higher background [3].
The key trade-off is that while increasing surface area boosts your signal, it can boost the background even more, potentially worsening the signal-to-background ratio. Furthermore, the total current (signal + background) may saturate your instrument's amplifiers if the electrode is too large, limiting usable electrode size and ultimate detection sensitivity [3].
Q2: What is amplifier saturation, and how can I prevent it?
Amplifier saturation (or clipping) occurs when the amplified signal exceeds the maximum input voltage range of your analog-to-digital converter (ADC). This causes irreversible distortion and loss of high-amplitude signal features [16]. In the context of an LNA, as it nears saturation, its gain reduces, weakening the signal of interest and increasing intermodulation products [19].
Prevention Strategies:
- Adjust Gain Settings: Reduce the amplifier's gain to ensure the output voltage does not exceed the ADC's input range [16].
- Use a Differential Potentiostat (DiffStat): By subtracting the capacitive background in hardware, the DiffStat outputs a signal that is predominantly faradaic current, preventing the large background from consuming the available dynamic range and causing saturation [3].
- Check Electrode Surface Area: If using a conventional potentiostat, a large electrode surface area can generate currents that are too high for the instrument's amplifiers. Using a smaller electrode may be necessary [3].
Q3: My electrode impedance is high. How will this impact my data quality?
High electrode impedance can meaningfully reduce data quality by increasing low-frequency noise, which decreases the signal-to-noise ratio (SNR) [20]. This is primarily because impedance imbalances between electrodes degrade the amplifier's common-mode rejection capability, making the system more susceptible to environmental noise [16] [20]. The consequence is that you will need to average more trials to achieve the same level of statistical significance in your averaged data, as the SNR of an average increases only with the square root of the number of trials [20].
Q4: Can I fix a signal-to-background problem solely through data analysis?
While digital background subtraction and filtering during data analysis can be effective, it is not the only or always the best solution. A key limitation of digital subtraction is that the large background currents remain in the raw data, which can still saturate instrument amplifiers and limit the use of larger, higher-signal electrodes [3]. Hardware-based solutions, like the differential potentiostat, subtract the background in real-time at the analog level, circumventing this limitation and often simplifying subsequent data processing [3].
Experimental Protocol: Suppressing Non-Faradaic Current with a DiffStat
This protocol outlines the methodology for using a differential potentiostat to suppress capacitive currents in a DNA-based hybridization assay, as described in the research [3].
Objective: To measure faradaic current from a methylene blue (MB)-tagged DNA target while suppressing the non-faradaic background.
Materials:
- Differential Potentiostat (DiffStat) [3]
- Two electrochemical cells, each with a Gold working electrode (W1 & W2)
- Split reference electrode (e.g., Ag/AgCl) and split counter electrode
- Thiolated DNA probe for self-assembled monolayers (SAM) on gold
- Target DNA sequence conjugated with Methylene Blue (MB-DNA)
- Control DNA sequence without the redox reporter (CTR-DNA)
Procedure:
- Electrode Preparation: Create DNA monolayers by immobilizing the thiolated DNA probe onto the gold working electrodes W1 and W2.
- Solution Addition:
- To the cell containing working electrode W1, add the MB-DNA target solution. This electrode will produce both faradaic and non-faradaic currents.
- To the cell containing working electrode W2, add the CTR-DNA control solution. This electrode, lacking the redox reporter, will produce only the non-faradaic background current.
- Electrochemical Connection: Connect the two cells using the split reference and counter electrodes to complete the electrochemical circuit while preventing cross-contamination.
- DiffStat Measurement: Run your chosen electrochemical technique (e.g., Chronoamperometry, Square-Wave Voltammetry). The DiffStat will simultaneously measure the current from W1 and W2 and output the analog-subtracted signal (W1 - W2), which is predominantly the faradaic current.
The following table summarizes key quantitative findings from the evaluation of a differential potentiostat (DiffStat) for background suppression [3].
| Parameter | Conventional Potentiostat | Differential Potentiostat (DiffStat) | Measurement Technique |
|---|---|---|---|
| Baseline Capacitive Current | Baseline level = 1 (reference) | ~5-fold suppression | Chronoamperometry (CA) |
| Accessible Electrode Size | Limited by amplifier saturation | Enables use of larger electrodes | N/A |
| Sensitivity Setting | Limited by high background | Enables higher sensitivity settings | N/A |
| Data Processing | Requires complex digital subtraction | Simplifies extraction of faradaic current | Square-Wave Voltammetry (SWV) |
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function / Rationale |
|---|---|
| Differential Potentiostat (DiffStat) | Core hardware for real-time, analog subtraction of non-faradaic background currents using two working electrodes [3]. |
| Gold Electrodes | Standard substrate for forming well-organized DNA self-assembled monolayers (SAMs) used in many modern bioassays [3]. |
| Methylene Blue (MB) | A common redox reporter molecule; its electron transfer efficiency can be optimized to improve the faradaic signal [3]. |
| Thiolated DNA Probes | Anchor onto gold electrodes to create a stable, functional sensing interface (e.g., for E-DNA or E-AB biosensors) [3]. |
| Control DNA (No Reporter) | Essential for the background (W2) electrode in a DiffStat setup to provide a matched non-faradaic current for subtraction [3]. |
Logical Workflow: Conventional vs. Differential Potentiostat Configurations
Troubleshooting Signal-to-Noise Ratio (SNR) Problems
Practical Strategies for Suppression: From Hardware Innovation to Measurement Techniques
Frequently Asked Questions (FAQs)
Q1: What is the primary advantage of using a DiffStat over a conventional potentiostat? The DiffStat provides order-of-magnitude improvements in sensitivity by suppressing non-Faradaic (capacitive) background currents through real-time analog subtraction. This allows the use of larger electrode surfaces and higher instrument sensitivity settings, which are often inaccessible with standard potentiostats due to amplifier saturation from high background currents [3].
Q2: My differential measurement shows an unstable baseline. What could be the cause? Unstable baselines are frequently caused by mismatched electrode surfaces or inconsistent monolayer coverage between the two working electrodes (W1 and W2). Ensure that both electrodes are prepared simultaneously using identical protocols for gold cleaning and DNA monolayer formation to guarantee equivalent capacitive backgrounds [3].
Q3: Can the DiffStat be used for "signal-off" assays? Yes, the DiffStat can uniquely convert traditional "signal-off" assays into "signal-on" formats. By configuring one electrode (W1) as the active sensor and the second (W2) with a non-responsive background, the differential output directly reports a positive signal upon analyte binding, simplifying data interpretation [3].
Q4: How does the DiffStat perform in complex biological matrices like serum? The differential measurement capability of the DiffStat enables effective background drift correction even in challenging environments like 50% human serum. The real-time subtraction compensates for drifting baselines caused by matrix effects, enhancing assay robustness [3].
Troubleshooting Guides
Problem: Excessive Noise in Differential Output
Symptoms: The differential signal is noisy, obscuring the faradaic response.
- Check 1: Verify the integrity of all physical connections to the two working electrodes, reference electrode, and counter electrode. Loose connections introduce significant noise.
- Check 2: Ensure both working electrodes (W1 and W2) are in separate electrochemical cells but share a common reference and counter electrode through a split configuration to prevent cross-contamination [3].
- Check 3: Confirm that the DNA monolayer coverages on W1 and W2 are nearly identical. Significant differences in surface properties lead to imperfect background subtraction.
Problem: Poor Suppression of Capacitive Current
Symptoms: The differential output still shows a significant sloping baseline in techniques like SWV.
- Check 1: The composition of the background solution in the W2 cell must be meticulously matched to the analytical solution in the W1 cell, including matching the ionic strength and the type and concentration of the DNA control sequence (e.g., CTR-DNA) [3].
- Check 2: The surface area of W1 and W2 must be as identical as possible. Even small differences in electrode geometry will result in residual uncompensated capacitive current.
Problem: Saturated Output Signal
Symptoms: The potentiostat's output is at its maximum voltage limit.
- Check 1: Reduce the gain or sensitivity setting on the DiffStat instrument. The much lower background current often allows for higher gain settings than with a ConStat, but the limit may be reached with very large electrodes.
- Check 2: If using a large surface area electrode, try a smaller one to reduce the absolute current magnitude, both faradaic and non-faradaic.
Experimental Protocols & Data
Key Experiment: Validation of Non-Faradaic Current Suppression
Objective: To demonstrate the effectiveness of the DiffStat in suppressing non-Faradaic current across different electrochemical techniques [3].
Methodology:
- Sensor Fabrication: Prepare two gold working electrodes (W1, W2) with thiolated DNA monolayers.
- Analyte Introduction: Introduce methylene-blue-tagged DNA (MB-DNA) target to the W1 cell and a control DNA (CTR-DNA) to the W2 cell.
- Electrochemical Measurement: Perform Chronoamperometry (CA), Cyclic Voltammetry (CV), and Square-Wave Voltammetry (SWV) measurements simultaneously with a conventional potentiostat (ConStat) and the DiffStat for comparison.
- Data Analysis: Compare the magnitude of the non-faradaic background and the signal-to-background ratio between the two instruments.
Key Results:
Table 1: Performance Comparison of ConStat vs. DiffStat [3]
| Electrochemical Technique | Non-Faradaic Current Suppression (DiffStat) | Signal-to-Background Improvement |
|---|---|---|
| Chronoamperometry (CA) | ~5-fold suppression of capacitive current | Significant increase |
| Cyclic Voltammetry (CV) | Significant suppression observed | Notable improvement |
| Square-Wave Voltammetry (SWV) | Background essentially reduced to zero | Dramatic improvement; simplifies data processing |
Workflow: DiffStat Operation for a DNA Hybridization Assay
The following diagram illustrates the experimental workflow and the core signaling principle of the differential potentiostat.
Protocol: Implementing Real-Time Background Correction in Serum
Objective: To leverage the DiffStat for continuous background drift correction in a complex matrix (50% human serum) [3].
Step-by-Step Procedure:
- Electrode Setup: Configure W1 as the biosensor electrode and W2 as a background electrode with a similar, non-specific DNA monolayer.
- Baseline Acquisition: Immerse both electrodes in 50% human serum and record a stable baseline using the DiffStat.
- Analyte Addition: Spit the target analyte into the solution containing W1.
- Continuous Monitoring: Record the differential output over time. The DiffStat will subtract the drifting background signal (common to both W1 and W2 in the serum matrix) from the specific faradaic signal generated at W1.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials and Reagents for DiffStat Experiments [3]
| Item | Function / Role in the Experiment |
|---|---|
| Gold Disk Electrodes | Serve as the platform for forming self-assembled monolayers (SAMs) and the DNA-based sensor interface. |
| Thiolated DNA Probes | Form the self-assembled monolayer on the gold electrode; provide the specific recognition element for the target. |
| Methylene Blue (MB) | A redox reporter molecule that is appended to DNA; generates the faradaic current measured in the assay. |
| Control DNA (CTR-DNA) | A non-redox-labeled DNA sequence used in the background electrode (W2) to match the chemical environment of W1. |
| Six-Chloride Iridium | Used as a split reference electrode, providing a stable and reproducible reference potential for both working electrodes. |
| Human Serum | A complex biological matrix used to validate assay performance and background correction in a clinically relevant medium. |
Fundamental Concepts: Why Use Dual Working Electrodes?
What is the primary advantage of using a dual working electrode system for background referencing?
The primary advantage is the significant suppression of non-Faradaic current, also known as capacitive or charging current. This current originates from the formation of an electrical double layer at the electrode-electrolyte interface and acts as a major interference background in electrochemical measurements. Unlike digital subtraction performed during data analysis, a dual-electrode system performs real-time analog subtraction of this background within the potentiostat hardware itself, leading to cleaner data and improved sensitivity [3].
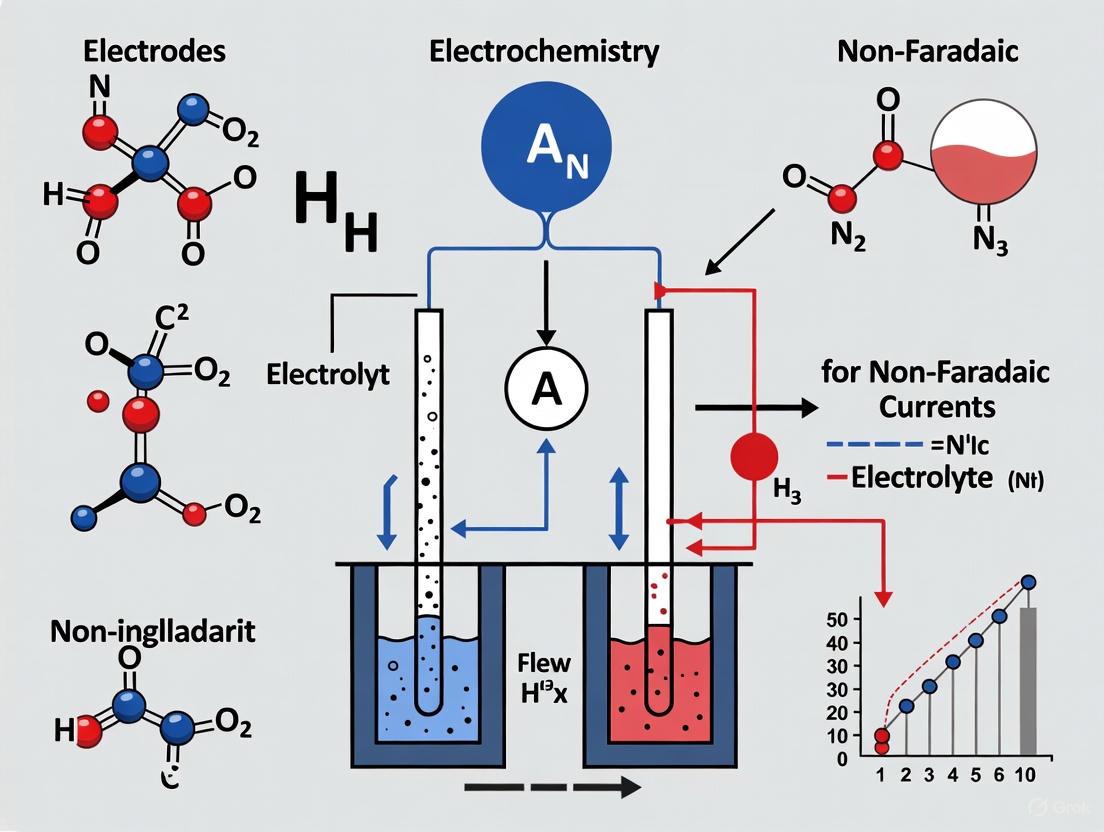
How does the differential potentiostat (DiffStat) configuration work?
A conventional potentiostat uses a single working electrode. In contrast, a differential potentiostat (DiffStat) utilizes a cell with two working electrodes (W1 and W2) [3]. Both electrodes are connected to matching current-to-voltage converter circuits. The signals from W1 (the experimental sensor) and W2 (the background reference) are collected simultaneously and fed into an on-board differential instrumentation amplifier, which performs continuous, analog subtraction of the W2 signal from the W1 signal. This process outputs a signal where the shared non-Faradaic background is greatly reduced, leaving a predominantly faradaic current [3].
The following diagram illustrates the signal flow and subtraction process in a DiffStat.
Experimental Protocols & Setups
Core Methodology: DNA Monolayer-Based Sensor
A validated application of this technology is for nucleic acid hybridization assays using DNA monolayers on gold electrodes [3].
- Sensor Design: A thiolated DNA probe is immobilized on a gold working electrode (W1) to form a self-assembled monolayer. The target is a complementary DNA strand labeled with a redox reporter, such as Methylene Blue (MB).
- Reference Electrode Design: The second working electrode (W2) is prepared identically but is exposed to a non-redox-active control DNA sequence (CTR-DNA). This ensures the non-faradaic background at both electrodes is nearly identical.
- Measurement: When the MB-DNA target hybridizes with the probe on W1, a faradaic current from the MB reporter is generated. The DiffStat subtracts the nearly identical capacitive background from W2, leaving a clean, background-suppressed faradaic signal from the hybridization event [3].
Electrode Configuration and Cell Setup
To prevent cross-contamination between the sensor and reference electrodes, it is recommended to fabricate W1 and W2 in two separate electrochemical cells. A split reference electrode and counter electrode can then be used to establish electrochemical contact with both cells simultaneously [3].
The table below summarizes the key techniques and their performance with a DiffStat.
Table 1: Performance of Differential Potentiostat Across Electrochemical Techniques
| Technique | Key Improvement with DiffStat | Observed Outcome |
|---|---|---|
| Chronoamperometry (CA) | Suppression of capacitance current in the baseline. | ~5-fold suppression of non-faradaic current was observed [3]. |
| Cyclic Voltammetry (CV) | Removal of the non-zero baseline. | Enables clearer visualization of faradaic peaks [3]. |
| Square-Wave Voltammetry (SWV) | Direct output of a background-subtracted signal. | Greatly simplifies data processing; non-faradaic current is suppressed "essentially to zero" [3]. |
Troubleshooting Common Issues
We observe excessive noise or signal drift after implementing the dual-electrode setup. What could be the cause?
- Mismatched Electrodes: The fundamental requirement for effective subtraction is that the non-faradaic background at W1 and W2 must be nearly identical. Ensure the two working electrodes are fabricated from the same material, have the same geometry and surface area, and are modified with the same monolayer chemistry and coverage.
- Reference Electrode Instability: A drifting reference electrode potential will skew the applied potential at both working electrodes, causing apparent signal drift. Test your reference electrode against a stable master reference electrode by measuring the open-circuit potential between them; a difference of >5 mV indicates a problem [21]. Always store reference electrodes in their proper filling solution to prevent crystallization and potential drift [21].
- Configuration Limitations: Be aware that using a two-electrode configuration (where one electrode acts as both reference and counter) or configurations where the counter electrode is a similar size to the working electrode can distort the electrochemical response and become the rate-limiting step. For benchtop validation, a standard three-electrode configuration with a large counter electrode is recommended [22].
Our faradaic signal is still low after background subtraction. How can we improve sensitivity?
The DiffStat's key benefit is enabling the use of larger electrode surface areas without amplifier saturation from high capacitive currents. Since faradaic current is also proportional to surface area, you can increase the surface area of your working electrodes to boost the absolute faradaic signal. The DiffStat will simultaneously handle the corresponding increase in non-faradaic current, which would otherwise be prohibitive on a standard potentiostat [3].
Can this system convert a "signal-off" assay into a "signal-on" assay?
Yes, this is a unique application. In a traditional signal-off assay, the binding of a target causes a decrease in signal. With a DiffStat, you can configure the system so that this decrease at W1 is subtracted from a stable background at W2, resulting in a net negative differential signal. By inverting this output, the assay is converted to a more intuitive signal-on format [3].
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 2: Key Reagents and Materials for Dual-WE Experiments
| Item | Function in the Experiment | Example / Specification |
|---|---|---|
| Differential Potentiostat (DiffStat) | Instrument that performs real-time analog subtraction of signals from two working electrodes. | Can be constructed from open-source designs [3]. |
| Paired Working Electrodes | The sensor (W1) and reference (W2) electrodes. Must be matched. | Gold disk electrodes; DNA-modified gold surfaces [3]. |
| Reference Electrode | Provides a stable, known potential for the electrochemical cell. | Ag/AgCl (e.g., in saturated KCl) [21] [23]. |
| Master Reference Electrode | A pristine reference electrode used solely to validate the stability of other reference electrodes. | A dedicated Ag/AgCl electrode stored in KCl solution [21]. |
| Counter Electrode | Completes the electrical circuit, often made from inert material. | Platinum wire or mesh [22] [23]. |
| Redox Reporter | Molecule that undergoes faradaic reaction, generating the analytical signal. | Methylene Blue (MB) [3]. |
| Surface Passivation Monolayer | Forms a well-defined interface on the electrode, reducing non-specific binding. | Thiolated DNA or alkanethiol self-assembled monolayers (SAMs) on gold [3]. |
The following workflow diagram outlines the key steps for setting up and running a successful experiment with dual working electrodes.
A fundamental challenge in electrochemical analysis is distinguishing the faradaic current, which arises from electron transfer in redox reactions, from the non-faradaic (capacitive) current, which originates from the charging and discharging of the electrical double layer at the electrode-electrolyte interface [24]. Non-faradaic currents act as a significant background interference, compromising assay sensitivity, limiting usable electrode surface area, and narrowing the detection range [3]. This technical brief provides troubleshooting guidance and methodologies for researchers to effectively suppress these capacitive currents, thereby enhancing the reliability of their electrochemical measurements.
Core Concepts & Troubleshooting FAQs
FAQ 1: What is the fundamental difference between Faradaic and Non-Faradaic currents?
- Faradaic Current: This is the current from actual redox reactions where electrons are transferred across the electrode-electrolyte interface, leading to chemical transformations. It is the analytical signal of interest that directly relates to the concentration of the analyte [24].
- Non-Faradaic Current (Capacitive Current): This current comes from the rearrangement of ions at the electrode surface to form an electrical double layer. No electron transfer occurs across the interface, and it does not cause a chemical change. It manifests as a time-dependent background signal that can obscure faradaic responses [3] [24].
FAQ 2: How can I experimentally identify if my signal is compromised by capacitive current?
Examine the raw output from techniques like chronoamperometry (CA) or square-wave voltammetry (SWV). A large, decaying transient in CA or a pronounced, sloping baseline in SWV that lacks the characteristic peak shape of a faradaic process strongly indicates significant capacitive interference [3].
FAQ 3: My data is noisy with a high background at larger electrode surfaces. What is the cause and solution?
Cause: The magnitude of the non-faradaic current is directly proportional to the electrode surface area (SA). Using larger electrodes to increase faradaic signal also amplifies the capacitive background, which can saturate instrument amplifiers and increase noise [3]. Solution: Consider a Differential Potentiostat (DiffStat) configuration. This hardware uses two working electrodes to subtract the capacitive background in real-time via analog circuitry, allowing the use of larger SAs and higher sensitivity settings without amplifier saturation [3].
Advanced Methodology: The Differential Potentiostat (DiffStat)
The DiffStat represents a hardware-level solution for non-faradaic current suppression. Its configuration and operation principle are as follows:
DiffStat Configuration and Workflow
Experimental Protocol for DiffStat Validation
This protocol outlines a hybridization-based DNA sensor assay to demonstrate the efficacy of the DiffStat in suppressing non-faradaic current [3].
1. Electrode and Cell Preparation:
- Working Electrodes (W1 & W2): Use two separate gold disk electrodes. Clean and polish them to a mirror finish. W1 and W2 must be fabricated in two separate electrochemical cells to prevent cross-contamination.
- Reference and Counter Electrodes: Employ a split reference electrode (e.g., Ag/AgCl) and a split counter electrode (e.g., Pt wire) to establish electrochemical contact with both cells simultaneously.
- DNA Monolayer Formation: Immerse both W1 and W2 in a solution of thiolated DNA probes to form a self-assembled monolayer (SAM) on each gold surface.
2. Analyte and Control Introduction:
- Working Electrode 1 (W1 - Signal): Introduce the target analyte, which is a complementary DNA strand labeled with a redox reporter (e.g., Methylene Blue, MB). This creates the MB-DNA complex, generating both faradaic and non-faradaic currents.
- Working Electrode 2 (W2 - Background): Introduce a control DNA sequence that is identical but lacks the Methylene Blue label (CTR-DNA). This carefully matches the non-faradaic components of W1 but provides no faradaic signal.
3. Electrochemical Measurement:
- Connect the prepared electrochemical cells to the DiffStat.
- Perform simultaneous measurements on W1 and W2 using Chronoamperometry (CA), Cyclic Voltammetry (CV), or Square-Wave Voltammetry (SWV).
- The DiffStat's internal differential amplifier performs real-time analog subtraction of W2's signal from W1's signal.
Performance Data and Analysis
Table 1: Quantitative Performance Comparison of Conventional vs. Differential Potentiostat
| Parameter | Conventional Potentiostat | Differential Potentiostat (DiffStat) | Improvement Factor |
|---|---|---|---|
| Capacitive Current Suppression | Baseline level | ~5-fold suppression in Chronoamperometry [3] | 5x |
| Faradaic Current | Unchanged | Unchanged [3] | - |
| Signal-to-Background Ratio | Limited by high background | Order-of-magnitude improvements [3] | >10x |
| Electrode Surface Area Use | Limited by amplifier saturation | Enables use of larger electrodes [3] | Expanded range |
| Data Processing for SWV | Requires complex digital subtraction | Simplified extraction of faradaic current [3] | Major simplification |
Research Reagent Solutions
Table 2: Essential Materials and Reagents for DNA-Based Electrochemical Assays
| Item | Function / Description | Example Application |
|---|---|---|
| Gold Disk Electrodes | Provides a stable, inert surface for forming DNA monolayers via gold-thiol chemistry. | Standard working electrode for DNA-based sensors [3]. |
| Thiolated DNA Probes | DNA strands with a thiol group at one terminus for covalent attachment to gold surfaces, forming the sensing monolayer. | Foundation for E-DNA, E-AB, and ECPA biosensors [3]. |
| Methylene Blue (MB) | A redox reporter molecule that can be appended to DNA. Its electron transfer generates the faradaic current. | Redox label in hybridization-based DNA sensors [3]. |
| Split Reference Electrode | A shared reference electrode (e.g., Ag/AgCl) with separate connections to maintain identical potential in both cells of a DiffStat setup. | Critical for the dual-cell DiffStat configuration [3]. |
Application Note: Converting Signal-OFF to Signal-ON Assays
A unique application of the DiffStat is its ability to convert traditional "signal-off" assays into more intuitive "signal-on" assays [3]. In a typical signal-off E-DNA sensor, target binding causes a decrease in the faradaic SWV peak. With the DiffStat:
- W1 is configured as the functional signal-off sensor.
- W2 is configured as a static, non-responsive DNA monolayer.
- When the target binds to W1, its signal decreases. The DiffStat subtracts the constant signal from W2, resulting in a differential output that increases as the signal from W1 decreases. This effectively converts the signal-off response into a signal-on output, which can be easier to interpret and quantify.
Frequently Asked Questions (FAQs)
Q1: What is the primary advantage of converting a signal-OFF assay to a signal-ON format? Converting a signal-OFF assay to a signal-ON format provides a direct, proportional relationship between the target analyte concentration and the reported signal. This significantly improves the assay's sensitivity and ease of interpretation, circumventing the inherent limitations of signal-OFF assays where signal suppression has a maximum limit of 100%, which can hinder detection of low analyte concentrations and make naked-eye observation difficult [25].
Q2: How does a differential potentiostat (DiffStat) correct for signal drift in complex samples like human serum? The DiffStat uses two working electrodes (W1 and W2) measured simultaneously. The background current (non-faradaic and drift components) from a "blank" electrode (W2) is subtracted in real-time via analog circuitry from the signal of the experimental electrode (W1). This hardware-level subtraction actively removes the background signal and its drift, which is crucial for accurate measurements in complex, variable matrices like 50% human serum [3].
Q3: Why is non-faradaic current a significant problem in electrochemical biosensors? Non-faradaic, or capacitive, current acts as a large, fluctuating background signal that can obscure the smaller faradaic current generated by the redox reaction of the reporter molecule. This high background limits the signal-to-noise ratio, narrows the detection range, and can saturate the instrument's amplifiers, thereby restricting the sensitivity and overall performance of the biosensor [3].
Q4: Can I use standard screen-printed electrodes (SPEs) with the DiffStat for on-site testing? The DiffStat configuration requires two working electrodes. While the cited research used custom-fabricated electrodes in separate cells to prevent cross-contamination, the principle is compatible with any two-electrode setup. For portability, a specially designed screen-printed electrode array that incorporates two working electrodes, a shared reference, and a shared counter electrode would be ideal and is a logical extension of this technology [3] [26].
Troubleshooting Guides
Problem 1: High Background Noise and Capacitive Current Overwhelming Faradaic Signal
Potential Causes and Solutions:
- Cause: Excessive electrode surface area leading to large capacitive currents.
- Solution: The DiffStat hardware is specifically designed to suppress this. If using a conventional potentiostat, you must reduce the electrode surface area, though this also reduces the faradaic signal. The DiffStat allows the use of larger electrodes for a stronger signal without the background penalty [3].
- Cause: Mismatched self-assembled monolayers (SAMs) or probe coverages on the two working electrodes (W1 and W2) in a DiffStat setup.
- Solution: Ensure that the fabrication of W1 and W2 is performed in parallel using identical protocols, solutions, and incubation times to guarantee that the non-faradaic background is nearly identical on both electrodes, enabling effective subtraction [3].
- Cause: Inadequate matching of the redox reporter and its control.
- Solution: When using a methylene blue (MB)-labeled DNA probe on W1, use an identical DNA sequence without the MB label on W2 (control electrode). This carefully matches the non-faradaic components between the two electrodes [3].
Problem 2: Successful Signal-OFF to Signal-ON Conversion but Poor Sensitivity
Potential Causes and Solutions:
- Cause: Inefficient displacement of the signaling probe in a competitive assay format.
- Cause: Suboptimal performance of the signaling element (e.g., nanozyme).
- Solution: If using a nanozyme (e.g., PVP-capped Pt nanocubes), ensure it is properly synthesized and that its activity is modulated effectively, for instance, with metal ions like Ag⁺, to achieve maximum catalytic signal amplification [25].
Problem 3: Signal Drift in Complex Biological Samples (e.g., Serum)
Potential Causes and Solutions:
- Cause: Non-specific adsorption of proteins or other matrix components onto the electrode surface.
- Solution: Employ a well-formed and dense self-assembled monolayer (SAM) to passivate the electrode surface. The DiffStat configuration provides continuous correction for this type of drift, as the non-specific adsorption will occur on both W1 and W2 and be subtracted out [3].
- Cause: Chemical or electrochemical fouling of the electrode.
- Solution: Use the DiffStat's real-time differential measurement for continuous background correction. This hardware approach circumvents the need for complex digital post-processing and provides stable baseline correction even in 50% human serum [3].
Experimental Protocols
Protocol 1: Converting a Signal-OFF DNA Hybridization Assay to Signal-ON Using a DiffStat
This protocol details the use of a differential potentiostat to transform a traditional signal-off sensor into a signal-on format [3].
1. Principle In a standard signal-off assay, target binding reduces the faradaic signal. The DiffStat uses two working electrodes: the experimental electrode (W1) with a signaling probe (e.g., MB-DNA), and a control electrode (W2) with a non-signaling probe (CTR-DNA). The analog subtraction of W2's signal from W1's signal cancels the common non-faradaic background. The faradaic signal from MB on W1 remains, converting a signal-off event on a single electrode (loss of MB signal) into a signal-on event in the differential readout (the background-subtracted signal is dominated by the faradaic current) [3].
2. Materials
- Differential Potentiostat (DiffStat): Custom-built hardware with two working electrode inputs and an onboard differential instrumentation amplifier [3].
- Working Electrodes (W1 & W2): Two gold electrodes fabricated in separate electrochemical cells to prevent cross-contamination.
- Reference and Counter Electrodes: A split reference electrode (e.g., Ag/AgCl) and counter electrode (e.g., Pt wire) for electrochemical contact with both cells.
- Probe Solutions:
- Thiolated DNA capture probe.
- Methylene Blue-labeled DNA target (MB-DNA) for W1.
- Identical, unlabeled DNA target (CTR-DNA) for W2.
- Buffer: Appropriate phosphate buffer or hybridization buffer.
3. Step-by-Step Procedure
| Step | Action | Critical Parameters |
|---|---|---|
| 1. | Electrode Preparation: Clean the gold working electrodes (W1 and W2) according to standard protocols (e.g., piranha treatment, electrochemical cycling). | Ensure identical surface roughness and cleanliness for both W1 and W2. |
| 2. | SAM Formation: Immobilize the thiolated DNA capture probe onto both W1 and W2 electrodes via self-assembly. | Use the same batch of probe solution and incubation time (often 1-24 hours) to achieve matched monolayer coverages. |
| 3. | Probe Introduction: To the cell containing W1, add the MB-DNA signaling probe. To the cell containing W2, add the CTR-DNA control probe. | The concentrations and sequences of MB-DNA and CTR-DNA must be identical except for the redox label. |
| 4. | Hybridization: Allow the labeled targets to hybridize with the surface-bound capture probes. | |
| 5. | DiffStat Measurement: Connect W1, W2, the reference, and counter electrodes to the DiffStat. Run the desired electrochemical technique (e.g., Square-Wave Voltammetry). | The DiffStat performs real-time analog subtraction of the W2 signal from the W1 signal. |
| 6. | Target Introduction (Signal-ON Readout): Introduce the unlabeled target analyte (e.g., complementary DNA). The analyte displaces the MB-DNA from W1, reducing its faradaic current, while the non-faradaic background on both electrodes remains matched. The differential output (W1 - W2) will show a net increase as the non-faradaic background is stripped away, revealing the signal-on response. |
4. Expected Results With a conventional potentiostat, target binding would cause a decrease in the MB faradaic peak (signal-OFF). With the DiffStat, the same binding event produces a differential signal where the background is effectively zero, and the displacement of MB leads to an apparent signal-ON response due to the removal of the suppressed background, simplifying data interpretation and improving the signal-to-noise ratio [3].
Protocol 2: Real-Time Background Drift Correction in 50% Human Serum
This protocol leverages the DiffStat for continuous measurement in complex biological fluids without complex data processing [3].
1. Principle The non-faradaic current and signal drift caused by matrix effects in human serum are similar on two identically prepared working electrodes. The DiffStat's synchronous measurement and analog subtraction of the signal from W2 (background electrode) from W1 (sensing electrode) removes these common-mode interferences in real-time, providing a stable baseline.
2. Materials
- Differential Potentiostat (DiffStat): As in Protocol 1.
- Electrodes: Same two-electrode setup as in Protocol 1.
- Biological Matrix: 50% human serum in an appropriate buffer.
3. Step-by-Step Procedure
| Step | Action | Critical Parameters |
|---|---|---|
| 1. | Sensor Preparation: Prepare the W1 and W2 electrodes identically, as described in Protocol 1, Steps 1-4. | The key is perfect matching of the two electrodes to ensure serum-induced drifts are identical. |
| 2. | Baseline in Buffer: Place a clean, blank buffer solution in both electrochemical cells. Record a baseline measurement with the DiffStat. | The differential signal should be a stable, flat line close to zero. |
| 3. | Introduction of Serum: Replace the buffer in both cells with 50% human serum. | Ensure both cells receive serum from the same stock solution at the same time. |
| 4. | Continuous Monitoring: Observe the differential output (W1 - W2) over time. The drift caused by the serum matrix will be subtracted, resulting in a stable baseline, allowing for accurate subsequent measurement of the target analyte. | This setup corrects for both non-faradaic charging current and low-frequency signal drift. |
4. Expected Results The DiffStat will output a significantly more stable baseline in 50% human serum compared to a conventional potentiostat. This stability allows for the direct and continuous monitoring of analyte binding without the need for digital background subtraction or frequent recalibration, facilitating adaptation to point-of-care testing [3].
Research Reagent Solutions
The following table lists key materials used in the advanced applications discussed.
| Item | Function/Application |
|---|---|
| Differential Potentiostat (DiffStat) | Core instrument that performs real-time analog subtraction of non-faradaic currents using two working electrodes [3]. |
| Methylene Blue (MB)-labeled DNA | Redox reporter molecule used as a signaling probe on the experimental working electrode (W1) [3]. |
| Unlabeled Control DNA (CTR-DNA) | Control probe, identical to the signaling probe but without the redox label, used on the background electrode (W2) to match non-faradaic components [3]. |
| Thiolated DNA Capture Probe | Forms a self-assembled monolayer (SAM) on gold electrodes, providing a well-defined surface for probe immobilization and hybridization [3]. |
| Gold Nanoparticles (AuNPs) | Used as electron-transfer mediators; can attach to captured bacteria to provide an electrical pathway across an insulating SAM, enhancing signal in impedimetric sensors [28]. |
| Pt-based Nanozymes (e.g., PVP-PtNC) | Artificial enzyme mimics with peroxidase-like activity; used in colorimetric assays to replace natural enzymes, offering higher stability and catalytic activity for signal amplification [25]. |
| Screen-Printed Electrode (SPE) Array | Provides a portable, disposable platform for electrochemical detection; can be designed with multiple working electrodes for differential measurements [26]. |
Visualized Workflows
Diagram 1: DiffStat Signal Conversion Logic
Diagram 2: Experimental Setup for Serum Drift Correction
Optimizing Assay Performance and Troubleshooting Common Pitfalls
Troubleshooting Guide: Frequently Asked Questions
FAQ 1: How can I reduce high non-Faradaic (capacitive) background currents that are interfering with the measurement of my faradaic signal?
- Issue: High capacitive background currents can overwhelm the analytical faradaic current, limiting signal-to-noise ratios and detection sensitivity, especially in complex matrices like serum or blood [3].
- Solution:
- Hardware Subtraction: Implement a differential potentiostat (DiffStat). This setup uses two working electrodes: one experimental (W1) and one as a background reference (W2). The circuitry performs real-time analog subtraction of the capacitive current from W2, outputting a signal predominantly composed of the faradaic current from W1 [3].
- Electrode Pretreatment: Perform electrochemical pretreatment cycles to reinforce the passivation layer on the electrode surface. This can help prevent electrolyte penetration and subsequent Faradaic side reactions that contribute to background and self-discharge [29].
- Optimize Electrode Area: While reducing the working electrode surface area can decrease capacitive current, it also reduces the faradaic signal. A DiffStat allows the use of larger electrodes without amplifier saturation, providing a better solution [3].
FAQ 2: My electrodeposited catalyst film is uneven. Which parameter is most critical to ensure uniform deposition?
- Issue: Uneven catalyst distribution on the substrate, often due to nucleation problems [30].
- Solution:
- Optimize Current Density: The applied current density during electrodeposition is a critical parameter. A current density that is too low or too high can lead to uneven, semi-continuous films. One study on Cu-based catalysts found that optimizing this parameter (e.g., 30 mA cm⁻²) was key to achieving uniform distribution and preventing pore blockage on gas diffusion layers [30].
- Control Catalyst Loading: Balance the current density with the total catalyst loading (e.g., 1 C cm⁻²). This balance prevents the formation of overly thick films that can block the electrode surface and harm performance [30].
FAQ 3: What is a systematic method to optimize multiple electrodeposition parameters simultaneously for a composite coating?
- Issue: The performance of a composite coating (e.g., Ni-Al₂O₃) depends on several interacting parameters, making one-factor-at-a-time optimization inefficient [31].
- Solution:
- Use the Taguchi Method: This statistical approach uses a designed orthogonal array (e.g., L16) to efficiently evaluate the effect of multiple factors with a minimal number of experiments [31].
- Key Parameters to Optimize: For Ni-Al₂O₃ composite coatings, the critical factors are current density (e.g., 2–5 A·dm⁻²), alumina concentration in the bath (e.g., 10–25 g·L⁻¹), deposition time (e.g., 15–60 min), and agitation rate (e.g., 200–350 rpm) [31]. The table below summarizes the optimization outcomes for these parameters.
| Factor | Levels Investigated | Primary Influence on Coating |
|---|---|---|
| Current Density | 2 to 5 A·dm⁻² | Affects grain refinement, incorporation of particles, and microhardness. |
| Al₂O₃ Concentration | 10 to 25 g·L⁻¹ | Directly influences the weight percentage of alumina incorporated into the nickel matrix. |
| Deposition Time | 15 to 60 min | Impacts coating thickness and crystallite size. |
| Agitation Rate | 200 to 350 rpm | Ensures uniform particle suspension and affects incorporation efficiency. |
| Optimization Outcome | Result | |
| Microhardness Increase | 164% improvement | |
| Al₂O₃ Incorporation | 400% rise | |
| Crystallite Size | Notable reduction |
Experimental Protocols
1. Synthesis of MoS₂/MWCNT Nanostructure:
- Materials: Functionalized multi-walled carbon nanotubes (MWCNTs-COOH), sodium molybdate dihydrate (Na₂MoO₄·2H₂O), thiourea, deionized water, and dimethylformamide (DMF).
- Procedure: a. Disperse 70 mg of MWCNTs-COOH in a 35 mL/35 mL mixture of deionized water and DMF. Ultrasonicate for 1.5 hours to create a well-dispersed suspension. b. Add 0.240 mmol of Na₂MoO₄·2H₂O and 0.912 mmol of thiourea to the MWCNT suspension under continuous stirring. c. Stir the mixture for 50 minutes at room temperature. d. Transfer the solution to a solvo/hydrothermal reactor and heat to synthesize the final MoS₂/MWCNT nanostructure.
2. Electrode Modification:
- Modify a commercial screen-printed carbon electrode (SPCE) by depositing the synthesized MoS₂/MWCNT nanostructure onto the working electrode surface.
3. Electrochemical Detection:
- Use techniques such as Differential Pulse Voltammetry (DPV) or Cyclic Voltammetry (CV) in a phosphate-buffered solution (PBS, 0.1 M, pH 2.0-9.0) to detect 4-Nitrophenol (4-NP).
- The sensor exhibits a linear range of 0.05 to 800.0 µM and a detection limit of 0.01 µM [32].
1. Optimization Procedure:
- System: Electrodeposition of Cu-based catalysts onto commercial GDLs to create Gas Diffusion Electrodes (GDEs).
- Key Parameters: Systematically vary the electrodeposition current density and the total charge passed (which determines catalyst loading).
- Performance Evaluation: Test the fabricated GDEs in a CO₂ electrolysis system at an applied current density of 200 mA cm⁻². Measure the Faradaic efficiency for valuable products like ethylene (C₂H₄).
2. Optimized Conditions and Outcome:
- Optimal Parameters: A current density of 30 mA cm⁻² and a charge density of 1 C cm⁻² (≈ 0.33 mg cm⁻² loading).
- Result: This combination achieved a high Faradaic efficiency (up to 70%) for C₂+ products while saving 50% catalyst material and 75% electrodeposition time compared to non-optimized parameters (e.g., 15 mA cm⁻² and 2 C cm⁻²) [30].
Research Reagent Solutions
Table 2: Essential Materials for Featured Experiments
| Item | Function / Application | Example Usage |
|---|---|---|
| Functionalized MWCNTs (MWCNTs-COOH) | Enhance electrical conductivity and provide a high-surface-area scaffold for composite nanostructures. | Used in the synthesis of MoS₂/MWCNT nanocomposites for sensor modification [32]. |
| Molybdenum Disulfide (MoS₂) | A semiconducting 2D material that provides catalytic active sites; often combined with conductive materials to overcome lower conductivity. | Combined with MWCNTs to create a high-performance voltammetric sensor for 4-Nitrophenol [32]. |
| Sodium Molybdate & Thiourea | Common precursors for the solvo/hydrothermal synthesis of MoS₂. | Used as Mo and S sources, respectively, in the synthesis of the MoS₂/MWCNT nanostructure [32]. |
| Watts-Type Bath (Ni Salts, H₃BO₃) | Standard electrolyte for nickel electrodeposition. The composition influences texture, microstructure, and properties. | Used as the base electrolyte for the electrodeposition of Ni-Al₂O₃ composite coatings [31]. |
| Alumina (Al₂O₃) Powder | A hard, inert particle used to create composite coatings, improving mechanical properties like microhardness and wear resistance. | Incorporated into a nickel matrix via co-electrodeposition [31]. |
| Tetraethylammonium tetrafluoroborate (TEABF₄) | A common salt used in organic electrolytes for electrochemical double-layer capacitors (EDLCs). | Used in acetonitrile to study self-discharge behavior and passivation layer effects in supercapacitors [29]. |
Experimental Workflow and Concept Diagrams
DiffStat Measurement Concept
Electrodeposition Optimization Workflow
In the context of research on correcting for non-Faradaic currents, managing the interplay between biosensors and their operating environment is paramount. Non-Faradaic processes, which involve capacitive charging and ion adsorption at the electrode-electrolyte interface without electron transfer, are highly susceptible to environmental factors such as pH fluctuations and material corrosion [33] [34]. These factors can significantly degrade signal integrity by contributing to non-specific adsorption (NSA), signal drift, and ultimately, inaccurate measurements [34]. This technical support center provides targeted troubleshooting guides and experimental protocols to help researchers maintain pH stability and corrosion resistance, thereby enhancing the reliability of biosensing platforms, particularly those utilizing sensitive electrochemical and electrochemical-surface plasmon resonance (EC-SPR) detection methods.
Fundamental Concepts: FAQs
FAQ 1: Why are pH fluctuations so detrimental to biosensors, especially in non-Faradaic measurements?
pH changes directly impact the electrochemical interface where non-Faradaic processes occur. A stable pH is crucial because:
- Surface Charge and Binding Affinity: The charge state of bioreceptors (e.g., antibodies, aptamers) and the sensor surface itself is pH-dependent. Fluctuations can alter the binding kinetics and efficiency of the target analyte [34].
- Non-Specific Adsorption (NSA): pH changes can promote the adsorption of non-target molecules (fouling) from complex samples like blood or serum. This fouling layer can mask the specific signal, increase background noise, and lead to false positives or negatives [34].
- Bioreceptor Stability: Many biological recognition elements can denature or lose activity outside their optimal pH range, leading to irreversible sensor degradation.
FAQ 2: How does sensor corrosion relate to signal instability and non-Faradaic effects?
Corrosion is an electrochemical process that directly compromises the sensor's physical and functional integrity:
- Degradation of the Sensing Interface: Corrosion damages the electrode surface, altering its capacitive properties which are central to non-Faradaic measurements [33]. This leads to signal drift and poor reproducibility.
- Leaching of Materials: Corroding electrodes can release ions into the sample, which may poison bioreceptors or catalyze undesirable side reactions, further confounding the signal [35].
- Loss of Biocompatibility: A corroding surface often becomes rougher and more prone to fouling, exacerbating issues with NSA [34].
FAQ 3: What is the relationship between non-Faradaic currents and the analytical signal in complex media?
In complex media, the signal has multiple components. Non-Faradaic processes are always present and are highly sensitive to the interface. In buffered, ideal solutions, this capacitive background may be stable. However, in real samples, fouling and environmental changes modify the interface capacitance, causing the non-Faradaic background to drift and distort the faradaic signal from the redox probe used for detection. Effectively, NSA manifests as an unstable non-Faradaic current, compromising the accuracy of the faradaic measurement [33] [34].
Troubleshooting Guides
Troubleshooting pH-Induced Signal Instability
| Symptom | Possible Cause | Solution | Verification Method |
|---|---|---|---|
| Signal drift in calibrators but stable in buffers | Inadequate buffer capacity for the sample matrix | Increase buffer concentration or switch to a buffer with a pKa closer to the target operational pH. | Test sensor in a sample with known analyte spiked into the actual matrix (e.g., serum). |
| Sudden, irreversible signal drop | Denaturation of pH-sensitive bioreceptor (e.g., antibody, enzyme) | Check the pH stability range of the bioreceptor and ensure storage and operational conditions are within limits. | Perform a functionality test with the bioreceptor in solution post-measurement. |
| High signal noise in complex samples (e.g., serum, milk) | NSA due to pH being near the isoelectric point of abundant proteins (e.g., albumin) | Adjust the operational pH to be away from the isoelectric point of major foulants. Implement an antifouling coating [34]. | Use EC-SPR to quantify the amount of protein adsorbed on the surface. |
| Inconsistent sensor response between batches | Variation in the local micro-environment pH due to different material lots or fabrication processes | Standardize sensor fabrication and preconditioning protocols. Incorporate a robust, solid-state reference electrode [36]. | Use a fluorescent pH dye to map the pH near the sensor surface. |
Troubleshooting Corrosion and Fouling
| Symptom | Possible Cause | Solution | Verification Method |
|---|---|---|---|
| Gradual, continuous signal drift over time | Progressive corrosion of electrode materials or formation of a fouling layer. | Use more inert electrode materials (e.g., gold, platinum). Apply conductive antifouling coatings like cross-linked protein films or hybrid materials [34]. | Monitor Open Circuit Potential (OCP) over time; a drifting OCP suggests surface corrosion. |
| Complete signal loss after exposure to aggressive media | Severe corrosion or passivation of the electrode surface. | Implement all-solid-state sensors designed for harsh environments [36] [35]. | Inspect the electrode surface using microscopy (SEM) post-measurement. |
| High background signal & reduced signal-to-noise ratio | Rapid fouling from sample matrix components (proteins, lipids). | Incorporate sample pre-treatment (dilution, filtration). Use coatings with tunable conductivity and thickness, such as new peptides or polymer layers [34]. | Measure charge-transfer resistance (Rct) via EIS; an increasing Rct indicates passivation. |
| Physical degradation of sensor (e.g., delamination) | Poor adhesion of functional layers exacerbated by electrochemical stress. | Optimize surface activation and functionalization chemistry (e.g., silanization, use of linkers). | Perform adhesion tests and visual inspection under a microscope. |
Experimental Protocols & Data Presentation
Protocol: Fabrication and Calibration of an All-Solid-State pH Sensor
This protocol is adapted from research on robust sensors for corrosive environments like concrete, making it highly suitable for ensuring pH stability in challenging biosensing applications [36] [35].
1. Objective: To fabricate and calibrate an all-solid-state pH sensor system consisting of an IrOx working electrode and a Mn/MnO2 reference electrode.
2. Materials (Research Reagent Solutions):
| Reagent/Material | Function |
|---|---|
| Iridium Wire (Ф 0.5 mm) | Substrate for the pH-sensitive working electrode. |
| Mn Powder & MnO2 Powder | Active components for the solid-state reference electrode. |
| Carbon Black Powder | Conductive additive for the reference electrode. |
| Polytetrafluoroethylene (PTFE) Resin | Binder for the reference electrode mixture. |
| IrCl₄, Oxalic Acid, H₂O₂, Na₂CO₃ | Chemicals for the carbonate melt oxidation synthesis of the IrOx film. |
| Epoxy Resin | Electrical insulation and physical protection of the sensor assembly. |
3. Step-by-Step Methodology:
Step 1: Fabrication of IrOx Working Electrode
- Prepare a growth solution by dissolving IrCl₄ in deionized water with oxalic acid as a complexing agent. Add H₂O₂ and adjust the pH to 10.5 with sodium carbonate [35].
- Using cyclic voltammetry, electrodeposit an IrOx film onto a pre-cleaned iridium wire by sweeping the potential between -0.3 V and +0.8 V (vs. a Red Rod reference) for 50 cycles at 50 mV/s [35].
Step 2: Fabrication of Mn/MnO2 Reference Electrode
- Mix Mn powder, MnO2 powder, and carbon black with a PTFE binder.
- Compact the mixture under high pressure to form a dense, homogeneous pellet [36].
Step 3: Sensor Assembly and Encapsulation
- Assemble the IrOx electrode and Mn/MnO2 pellet into a single probe body.
- Encapsulate the assembly with epoxy resin, ensuring only the active sensing surfaces are exposed [36].
Step 4: Calibration
- Immerse the sensor in a series of standard pH buffer solutions (e.g., pH 2 to 13).
- Measure the open-circuit potential (OCP) of the IrOx electrode against the Mn/MnO2 reference for each buffer.
- Plot the potential (E) vs. pH to obtain a calibration curve (Nernstian slope of -59.16 mV/pH at 25°C indicates ideal performance).
4. Data Interpretation:
- A linear potential-pH response with a slope close to the theoretical Nernstian value confirms sensor functionality.
- Low hysteresis and fast response time (<30 seconds) are indicators of a robust sensor.
- Long-term stability can be verified by repeated calibrations over days or weeks, showing minimal drift in the calibration slope or intercept [36].
Quantitative Performance of Sensor Materials
Table 1: Performance characteristics of advanced pH sensor materials reported in recent literature.
| Sensor Material | Fabrication Method | Linear Range (pH) | Sensitivity (mV/pH) | Stability & Notes | Source |
|---|---|---|---|---|---|
| IrOx Electrode | Carbonate Melt Oxidation | 2 - 13 | ~ -59.2 (Nernstian) | Remarkable long-term stability, ideal for embedded use in harsh (e.g., mortar) environments. | [36] |
| IrOx Electrode | Electrodeposition (Yamanaka method) | 2 - 13 | ~ -59.2 (Nernstian) | Robust, fast response. Used for monitoring sulfuric acid attack in mortars. | [35] |
| Optical Nanosensor | Nanoparticle "Tattoo" (Ionophore-based) | Physiological range | Colorimetric | For subcutaneous monitoring. Changes color based on ion concentration (K+, Na+, etc.). | [37] |
Table 2: Efficacy of selected antifouling coatings for electrochemical (EC) biosensors.
| Antifouling Coating | Material Type | Target Sample | Key Performance Highlight | Source |
|---|---|---|---|---|
| Cross-linked Protein Films | Biological | Serum, Blood | Forms a dense, hydrophilic barrier that resists protein adsorption. | [34] |
| New Peptides | Biological | Complex Media | Engineered sequences that minimize hydrophobic and electrostatic interactions with foulants. | [34] |
| Hybrid Materials | Synthetic/Biological Composite | Milk, Serum | Combines the conductivity of nanomaterials with the antifouling properties of polymers. | [34] |
The Scientist's Toolkit: Diagrams and Workflows
Signaling Pathway: Impact of Environmental Factors on Biosensor Signal
This diagram visualizes the logical relationship between environmental stressors, their physical effects on the sensor, and the subsequent impact on the analytical signal, within the context of non-Faradaic processes.
Environmental Stressors Impact on Signal
Experimental Workflow: Validating Sensor Stability and Antifouling Performance
This workflow outlines a systematic procedure for testing and validating the stability of a biosensor against pH changes, corrosion, and fouling.
Sensor Stability Validation Workflow
FAQs and Troubleshooting Guides
Frequently Asked Questions (FAQs)
Q1: What is the primary cause of high background interference in electrochemical assays, and how can it be mitigated? High background interference, specifically non-faradaic (capacitive) current, is a key sensitivity-limiting factor in electrochemical assays, particularly those using DNA monolayers on gold electrodes [3]. This capacitive current originates from the formation of an electrical double layer at the electrode-monolayer surface and acts as a non-zero baseline, limiting the signal-to-noise ratio and detection range [3]. Mitigation Strategy: A primary hardware-based solution is the use of a differential potentiostat (DiffStat). This system utilizes two working electrodes: an experimental electrode (W1) and a background electrode (W2). The signals from both electrodes are collected simultaneously, and the background current from W2 is analog-subtracted from the signal of W1 in real-time, outputting a signal predominantly composed of the faradaic current [3].
Q2: How does data acquisition frequency (sampling rate) impact my measurement, and how do I select the correct rate? The data acquisition rate, or sampling rate, defines how many data points are collected per second and is fundamental to capturing your signal accurately [38].
- Impact of a Low Rate: Too slow a sampling rate risks undersampling, which can cause aliasing (where high-frequency signals are misrepresented as lower frequencies) and lead to a loss of critical signal detail [38].
- Impact of a High Rate: While a fast rate captures more detail, it generates large data volumes, demanding significant storage and processing power, which can be inefficient for long-term monitoring [38].
- Selection Guideline: Follow the Nyquist Theorem, which states the sampling rate must be at least twice the highest frequency present in the signal you wish to measure [38]. For practical purposes, aim for a rate that captures 25-50 points across the narrowest peak in your analysis [39].
Q3: My potentiostat has different sensitivity settings (e.g., High, Medium, Low). What do these control? In instruments like potentiostats, sensitivity settings often control how the instrument's amplifiers respond to the measured current. A higher sensitivity setting makes the instrument more responsive to small current changes but also more susceptible to amplifier saturation from large currents (both faradaic and non-faradaic) [3]. Using hardware background suppression, like a DiffStat, allows you to use higher sensitivity settings and larger electrodes without saturation, enabling order-of-magnitude improvements in sensitivity [3].
Troubleshooting Common Experimental Issues
Problem 1: Excessive Capacitive Background in Voltammetric Measurements
- Symptoms: A large, sloping baseline in cyclic voltammetry (CV) or a high, non-faradaic background current in square-wave voltammetry (SWV) that obscures the faradaic peaks [3].
- Possible Causes and Solutions:
- Cause: Large electrode surface area amplifying both faradaic and non-faradaic currents [3].
- Solution: If a differential potentiostat is not available, reduce the working electrode's surface area. Be aware this will also reduce your faradaic signal [3].
- Cause: Hardware limitation leading to saturation at high-sensitivity settings [3].
- Solution: Implement a differential measurement approach. Use two matched working electrodes: one with your sensing layer (W1) and a "blank" with just the monolayer (W2). Perform sequential measurements and digitally subtract the background. While not as effective as real-time analog subtraction, this can still reduce background [3].
Problem 2: Poor Signal-to-Noise Ratio (S/N) Despite a Strong Signal
- Symptoms: A detectable signal is present, but it is noisy, making precise quantification difficult.
- Possible Causes and Solutions:
- Cause: Inappropriate data acquisition rate or filtering [39].
- Solution: Optimize detector parameters. As demonstrated in HPLC, a slower data rate combined with a slower filter time constant (a noise filter that removes high-frequency noise) can significantly reduce baseline noise and improve the S/N ratio [39].
- Cause: Electrical noise from the environment.
- Solution: Ensure all instrumentation is properly grounded and use shielded cables. Conduct experiments within a Faraday cage if possible.
Problem 3: Unstable Sampling Rate During Long-Term Data Acquisition
- Symptoms: Inconsistent time intervals between data points during long recordings, leading to data distortion.
- Possible Causes and Solutions:
- Cause: Inefficient data acquisition software or hardware limitations, especially in custom-built, low-cost DAQ systems [40].
- Solution: For software, optimize the code structure. Changing the data type from a high-precision "Double" to a more efficient type like "uint" can increase the maximum stable sampling rate. For hardware, ensure a stable and adequate power supply, as its quality can directly influence DAQ performance [40].
Table 1: Guide to Optimizing Data Acquisition Parameters
| Parameter | Definition | Optimization Guideline | Impact on Measurement |
|---|---|---|---|
| Sampling Rate | Number of samples taken per second (Hz) [38]. | At least 2x the highest signal frequency (Nyquist Theorem); 25-50 points across the narrowest peak [39] [38]. | Too low: Aliasing, loss of detail. Too high: Large file sizes, inefficient [38]. |
| Filter Time Constant | A noise filter that removes high-frequency noise [39]. | Use a slower time constant to reduce baseline noise; balance with acceptable peak broadening [39]. | Slower constant: Less noise, broader peaks. Faster constant: More noise, sharper peaks [39]. |
| Sensitivity (Gain) | Amplification of the input signal. | Use the highest setting that does not cause amplifier saturation. Background suppression enables higher gain [3]. | Higher sensitivity: Better detection of weak signals, risk of saturation. Lower sensitivity: Prevents saturation, may miss weak signals [3]. |
Table 2: Research Reagent Solutions for DNA-Based Electrochemical Sensing
| Reagent/Material | Function in Experiment | Example Context |
|---|---|---|
| Gold Electrode | A common working electrode platform for forming self-assembled monolayers (SAMs) [3]. | Used as a base for thiolated DNA probe immobilization in E-DNA and E-AB biosensors [3]. |
| Thiolated DNA Probes | Forms a dense, organized monolayer on the gold surface, serving as the recognition element [3]. | The foundation for sensors that can detect nucleic acid hybridization or specific proteins [3]. |
| Methylene Blue (MB) | A redox reporter molecule that undergoes electron transfer at the electrode surface [3]. | Appended to DNA sequences; a change in its electron transfer efficiency signals a binding event [3]. |
| Reference Electrode (e.g., Ag/AgCl) | Provides a stable, known potential against which the working electrode is controlled [41]. | An essential component of the standard three-electrode electrochemical cell [41]. |
| Counter/Auxiliary Electrode (e.g., Pt wire) | Completes the electrical circuit, allowing current to flow [41]. | An essential component of the standard three-electrode electrochemical cell [41]. |
Experimental Protocols
Protocol 1: Implementing a Differential Measurement for Background Subtraction
This protocol outlines the methodology for using a differential potentiostat (DiffStat) to suppress non-faradaic currents, as described in [3].
1. Objective: To acquire electrochemical signals with suppressed capacitive background using a two-working-electrode differential configuration. 2. Materials: * Differential Potentiostat (DiffStat) * Two Gold working electrodes (W1 and W2) * Shared Reference Electrode (e.g., Ag/AgCl) and Counter Electrode * Electrolyte solution * Thiolated DNA probes and target analytes 3. Methodology: * Step 1: Electrode Preparation. Prepare two gold working electrodes. On W1 (signal electrode), immobilize the DNA-based sensing layer. On W2 (background electrode), immobilize an identical but non-signaling monolayer (e.g., without a redox reporter or specific recognition element). * Step 2: Cell Setup. Place both electrodes in the electrochemical cell containing the electrolyte. To prevent cross-contamination, W1 and W2 can be fabricated in separate cells connected via a split reference and counter electrode [3]. * Step 3: Instrument Connection. Connect W1 and W2 to the DiffStat's respective working electrode inputs. Connect the shared reference and counter electrodes. * Step 4: Data Acquisition. Run your electrochemical technique (e.g., SWV, CA, CV). The DiffStat will automatically and synchronously subtract the current from W2 from the current from W1 during the current-to-voltage conversion. * Step 5: Output. The DiffStat outputs a signal where the non-faradaic background is significantly suppressed, revealing the faradaic current.
Protocol 2: Optimizing Detector Parameters for Improved S/N
Adapted from PDA detector optimization [39], this logic can be applied to various analytical instruments.
1. Objective: To systematically optimize detector parameters to maximize the signal-to-noise ratio (S/N). 2. Methodology: * Step 1: Establish a Baseline. Analyze your sample using the instrument's default parameters and record the S/N. * Step 2: Optimize Data Rate. Inject the sample at different data rates (e.g., 1, 2, 10, 40 Hz). Select the rate that provides sufficient points across your peak (e.g., 25-50) without introducing excessive noise [39]. * Step 3: Optimize Filtering. With the optimal data rate, test different filter time constants (e.g., No filter, Fast, Normal, Slow). A slower filter typically reduces noise but may broaden peaks; select the setting that gives the best S/N [39]. * Step 4: Advanced Optimization. If available, explore features like absorbance compensation (in optical detectors) which subtracts noise from a non-absorbing wavelength region, or adjust slit widths/resolution to balance light throughput and spectral fidelity [39].
Conceptual and Workflow Diagrams
Diagram 1: Differential vs. Conventional Potentiostat Configuration. The DiffStat uses two working electrodes and real-time analog subtraction to suppress non-faradaic background [3].
Diagram 2: Workflow for Parameter Optimization. A systematic iterative process for optimizing data acquisition frequency and sensitivity to minimize background noise.
Addressing Fouling and Drift in Complex Biological Matrices like Whole Blood
Technical Support Center: Troubleshooting Guide
This technical support center provides targeted solutions for researchers combating fouling and signal drift in electrochemical biosensors operating in complex biological matrices such as whole blood. These challenges are particularly critical in the context of correcting for non-Faradaic currents, as biofouling layers and signal instability can severely compromise measurement accuracy by contributing to background interference and reducing signal-to-noise ratios. The following guides and protocols address these issues through advanced materials science, innovative instrumentation, and data processing techniques.
Troubleshooting FAQs
1. My sensor signal degrades rapidly when exposed to whole blood. What antifouling strategies can I implement? Biofouling from proteins and cells in whole blood rapidly degrades sensor performance. Implement these surface modification strategies:
- Zwitterionic Polymer Coatings: Graft poly-sulfobetaine methacrylate (SBMA) and polydopamine (PDA) composites onto electrode surfaces. This creates a durable, hydrophilic barrier that resists protein adsorption and cell adhesion, significantly reducing signal decay. The coating demonstrates high robustness to pH, temperature, and mechanical stress variations [42].
- Cross-linked Protein Albumin Coatings: Apply a simple, ultrafast (<1 minute) dip-coating method using cross-linked bovine serum albumin (BSA) infused with conductive, pentaamine-functionalized graphene particles. This coating provides exceptional antifouling properties and can be stored at room temperature for at least 20 weeks while maintaining electrode conductivity for up to 9 weeks in unprocessed biological samples [43].
- Peptide-based Antifouling Layers: Construct antifouling surfaces with specific peptide molecules on a conductive metal-organic framework (MOF). This combination ensures high sensitivity even in complex serum matrices by preventing non-specific binding while maintaining efficient electron transfer [44].
2. How can I minimize the impact of non-Faradaic (capacitive) currents in my measurements? Non-Faradaic currents act as a significant background interference, limiting sensitivity. Suppress them through these methods:
- Differential Potentiostat (DiffStat) Hardware: Utilize a potentiostat with two working electrodes (W1 for signal, W2 for background) and integrated differential operational amplifiers. This setup performs real-time analog subtraction of capacitive currents during measurement, before digital conversion. This hardware approach suppresses non-faradaic background, allows use of larger electrode surface areas, and enables higher sensitivity settings without amplifier saturation [3].
- Electrode Design and Data Processing: Optimize self-assembled monolayer (SAM) coverage and electrode surface area. Combine this with digital background subtraction techniques in square-wave voltammetry (SWV) or pulse voltammetry. While software-based, this can effectively isolate the faradaic current component during data analysis [3].
3. My sensor exhibits significant signal drift over time in continuous monitoring. How can I correct for this? Long-term drift, often from sensor aging or environmental changes, requires correction strategies:
- Empirical Unsupervised Drift Correction: For electrochemical gas sensors, a linear correction model can be applied. First, establish a baseline calibration using Multiple Linear Regression that accounts for temperature and humidity. Then, correct for long-term aging drift by using Particle Swarm Optimization (PSO) to identify the parameters of a correction model that compensates for changes in sensor sensitivity and baseline over time. This method can maintain accurate concentration estimation for at least 3 consecutive months without needing recalibration with labeled data [45].
- Stable Antifouling Interfaces: As described in FAQ #1, implementing robust antifouling coatings like SBMA@PDA directly reduces a major source of signal drift by preventing the buildup of fouling layers that alter the electrode-solution interface [42].
4. Can I detect low-concentration biomarkers directly in whole blood without sample preprocessing? Yes, with optimized sensor platforms. A multiplexed platform using the BSA-graphene coating mentioned above has demonstrated single-digit picogram per milliliter (pg/mL) sensitivity for clinically relevant biomarkers (e.g., for myocardial infarction and traumatic brain injury) in unprocessed human plasma and whole blood. This sensitivity is achieved within minutes and is at least 50 times more sensitive than traditional ELISA. The signal remains stable enough to be measured after one week of storage [43].
Experimental Protocols for Key Methodologies
Protocol 1: Applying an Ultrarapid Antifouling Conductive Nanomaterial Coating This protocol is adapted from the method demonstrating highly sensitive multiplexed detection in whole blood [43].
- Objective: To create a stable, conductive, antifouling layer on an electrochemical sensor for direct use in complex biofluids.
- Materials:
- Electrochemical sensor chips
- Bovine Serum Albumin (BSA)
- Pentaamine-functionalized graphene particles
- Cross-linking agent (e.g., glutaraldehyde)
- Dip-coating apparatus
- Rapid heating source (e.g., hot plate or oven)
- Procedure:
- Prepare a solution of BSA and pentaamine-functionalized graphene particles in a suitable buffer.
- Activate the sensor chip surface with a primer layer if necessary.
- Dip the sensor chip into the BSA-graphene solution for a few seconds to ensure complete coverage.
- Immediately transfer the chip to a pre-heated plate or oven for rapid thermal cross-linking (< 1 minute total processing time).
- Allow the coated sensor to cool and stabilize. The coated sensor can be stored at room temperature.
- Validation: Test coating stability and conductivity by running cyclic voltammetry in a standard redox probe solution (e.g., Ferri/Ferrocyanide) over time. Validate antifouling performance by measuring the signal response for a target biomarker in undiluted whole blood and comparing the signal stability and noise to an uncoated sensor.
Protocol 2: Implementing a Differential Potentiostat (DiffStat) for Non-Faradaic Current Suppression This protocol is based on hardware developed for DNA-based sensors [3].
- Objective: To suppress capacitive background currents in real-time using analog circuitry.
- Materials:
- Open-source potentiostat platform (e.g., DStat)
- Components for two transimpedance amplifiers (TIAs)
- Differential instrumentation amplifier
- Electrochemical cell with two working electrodes (W1, W2), a shared reference electrode (split), and a shared counter electrode.
- Procedure:
- Circuit Modification: Modify a standard potentiostat circuit to incorporate a second, identical channel for the second working electrode (W2). The currents from both working electrodes (W1 and W2) are converted to voltage by two separate but matched TIAs.
- Differential Subtraction: Feed the outputs of the two TIAs into a differential instrumentation amplifier. This stage performs real-time analog subtraction of the signal from W2 (background) from the signal from W1 (analyte).
- Electrode Preparation: Functionalize the first working electrode (W1) with the sensing element (e.g., aptamer, antibody). Prepare the second working electrode (W2) identically but without the signaling component (e.g., using a control DNA sequence without a redox reporter) to match its capacitive properties to W1.
- Measurement: Run the electrochemical technique (e.g., Chronoamperometry, SWV) as usual. The output from the DiffStat will be the differential current, which is predominantly faradaic.
- Validation: Compare cyclic voltammograms or square-wave voltammograms of a redox reporter (e.g., Methylene Blue) obtained with the DiffStat and a conventional potentiostat. The DiffStat should show a significantly flattened baseline (near-zero non-faradaic current) while retaining the characteristic faradaic peaks.
Performance Data and Material Solutions
Table 1: Comparison of Antifouling and Drift-Correction Strategies
| Strategy | Core Mechanism | Key Performance Metrics | Best For |
|---|---|---|---|
| Zwitterionic Coating (SBMA@PDA) [42] | Creates a hydrophilic, protein-repellent surface. | Reduces signal drift; robust to pH/temperature stress; enables drug detection in artificial interstitial fluid. | Wearable, continuous sensors in variable environments. |
| BSA-Graphene Coating [43] | Forms a cross-linked, conductive antifouling layer. | Single-digit pg/mL sensitivity in whole blood; stable signal for 1 week; <1 min coating time. | High-sensitivity, multiplexed biomarker detection in raw samples. |
| Differential Potentiostat (DiffStat) [3] | Real-time analog subtraction of capacitive current. | Order-of-magnitude sensitivity improvement; enables use of larger electrodes. | Assays with high non-faradaic background, point-of-care applications. |
| MOF-enhanced Immunoprobe [44] | Oriented antibody immobilization on antifouling MOF. | Detection limit of 5 pg/mL for cancer markers in serum. | Highly specific clinical biomarker detection in blood. |
| Empirical Drift Correction [45] | Algorithmic correction of aging drift using PSO. | Maintains accuracy for 3+ months without recalibration. | Long-term environmental gas monitoring. |
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Materials for Fouling- and Drift-Resistant Sensors
| Item | Function | Example & Notes |
|---|---|---|
| Zwitterionic Monomer | Forms a highly hydrophilic, neutrally-charged antifouling polymer layer. | Sulfobetaine methacrylate (SBMA). Used with polydopamine (PDA) for robust coating adhesion [42]. |
| Conductive Nanomaterial | Enhances electron transfer in thick antifouling layers and improves sensitivity. | Pentaamine-functionalized graphene. Infused into BSA matrix to create a conductive antifouling composite [43]. |
| 2D Metal-Organic Framework (MOF) | Provides a high-surface-area platform for oriented antibody immobilization and enhanced electron transfer. | Zn-TCPP nanosheets on graphene oxide. The unsaturated Zn sites allow for oriented binding of biomolecules [44]. |
| Differential Potentiostat | Hardware for real-time suppression of non-faradaic/capacitive currents. | DiffStat. An open-source design can be built to utilize two working electrodes for background subtraction [3]. |
Diagnostic and Workflow Visualizations
Diagram 1: A diagnostic workflow for identifying sensor issues and selecting appropriate mitigation strategies.
Diagram 2: The signal path and working principle of a Differential Potentiostat (DiffStat) for real-time non-Faradaic current suppression.
Validating Performance and Comparing Method Efficacy for Robust Assays
Troubleshooting Guides
Guide 1: Diagnosing and Resolving Incomplete Non-Faradaic Current Suppression
Problem: Your electrochemical measurement shows a high, sloping baseline, indicating persistent non-Faradaic (capacitive) current interference after applying background subtraction.
Check Your Electrode Pair Matching (Analog Subtraction)
- Symptom: High residual background in differential potentiostat (DiffStat) measurements.
- Cause: The two working electrodes (W1-signal and W2-background) have mismatched surface areas, monolayer coverages, or double-layer capacitances [3].
- Solution: Pre-qualify electrode pairs by running cyclic voltammetry in a blank electrolyte solution. Select pairs where the background currents differ by less than 5%. Ensure identical fabrication, cleaning, and DNA monolayer formation protocols for both electrodes [3].
Verify Subtraction Timing (Digital Subtraction)
- Symptom: Noisy or distorted baseline in digitally subtracted square-wave voltammetry (SWV).
- Cause: The background scan used for digital subtraction was collected at a different time than the analytical scan, and experimental conditions (e.g., temperature, buffer composition) drifted [46].
- Solution: For digital methods, minimize the time interval between collecting the background and analytical scans. Use an average of multiple background scans collected immediately before and after the analyte introduction to compensate for linear drift [46].
Assess Data Processing Parameters
- Symptom: The faradaic peak is distorted after digital processing.
- Cause: Overly aggressive smoothing or incorrect baseline fitting models.
- Solution: Apply mild smoothing filters (e.g., Savitzky-Golay) and use a baseline correction model that fits the non-faradaic current shape in your specific technique (e.g., decaying exponential for chronoamperometry) [47].
Guide 2: Addressing Poor Signal-to-Noise Ratio After Subtraction
Problem: After background subtraction, the faradaic signal is weak, noisy, or indistinguishable from the baseline.
Increase Signal Averaging (Digital Subtraction)
- Cause: The inherent noise of the potentiostat amplifier is large compared to the small faradaic current.
- Solution: For digital methods, increase the number of scans (n) that are averaged. The signal-to-noise ratio (SNR) improves with √n. This is a standard capability in most modern potentiostats [46].
Utilize Higher Sensitivity Settings (Analog Subtraction)
- Cause: The instrument's gain is limited to avoid amplifier saturation from the large initial non-faradaic current.
- Solution: The DiffStat analog subtraction removes most of the capacitive current before the first amplifier stage. This allows you to safely increase the potentiostat's sensitivity (gain) by an order of magnitude, boosting the faradaic signal without saturation [3] [48].
Optimize Electrochemical Technique Parameters
- Cause: Non-optimal pulse times, frequencies, or step potentials that do not allow sufficient decay of non-faradaic currents relative to faradaic currents.
- Solution: In SWV, increase the pulse time. In chronoamperometry, increase the delay before starting current measurement. This allows more time for the capacitive current to decay exponentially [49].
Frequently Asked Questions (FAQs)
General Concepts
Q1: What is the fundamental difference between digital and analog subtraction of non-Faradaic currents?
- Analog Subtraction is a hardware-based method. It uses a differential potentiostat (DiffStat) with two working electrodes to perform real-time, continuous subtraction of background currents within the analog circuitry, before analog-to-digital conversion [3] [48]. The output signal is already background-corrected.
- Digital Subtraction is a software-based method. It involves sequentially collecting a background scan (without analyte) and an analytical scan (with analyte) using a standard potentiostat. The background is then digitally subtracted from the analytical scan during data analysis [46].
Q2: Why is suppressing non-Faradaic current so critical in electrochemical biosensing?
Non-faradaic (capacitive) current arises from the charging and discharging of the electrical double layer at the electrode-solution interface. It does not involve electron transfer from redox reactions (the faradaic process) [50]. This capacitive current can be orders of magnitude larger than the faradaic current from your target analyte, acting as a large, interfering background. Effective suppression is essential for:
- Achieving low detection limits and high sensitivity [3] [47].
- Enabling the use of larger electrode surfaces for greater total signal [3].
- Simplifying data interpretation by revealing the true faradaic signal [48].
Method Selection & Comparison
Q3: When should I choose analog subtraction over digital subtraction?
Refer to the following table for a direct comparison to guide your choice.
| Parameter | Analog Subtraction (DiffStat) | Digital Subtraction |
|---|---|---|
| Principle | Real-time hardware subtraction using two working electrodes [3] | Software-based subtraction of sequentially acquired scans [46] |
| Best For | Complex matrices (e.g., serum, blood) [3], real-time monitoring, point-of-care applications [48] | Controlled lab environments, well-characterized systems, when electrode pairing is impractical |
| Key Advantage | Enables use of larger electrodes & higher gain; continuous drift correction [3] | No specialized hardware needed; leverages standard potentiostat software |
| Throughput | Higher (background correction is simultaneous) | Lower (requires separate background scan) |
| Limitation | Requires a matched pair of working electrodes [3] | Vulnerable to background drift between scans [46] |
Q4: Can these methods convert a "signal-off" assay into a "signal-on" assay?
Yes, this is a unique capability of the analog subtraction approach. In a traditional "signal-off" assay, the faradaic current decreases upon target binding. With a DiffStat, you can configure the experiment so that this decrease at the sensor electrode (W1) is measured relative to a stable background electrode (W2). The differential output (W1 - W2) will show a negative current peak. By inverting the potentiostat leads, this negative peak is presented as a positive "signal-on" output, which is often more intuitive and reliable for quantification [3].
Experimental Implementation
Q5: What are the essential reagents and materials for implementing these methods in a DNA-based sensor?
The table below lists key items for a typical experiment as described in the literature [3].
| Research Reagent Solution | Function in the Experiment |
|---|---|
| Thiolated DNA Probes | Forms a self-assembled monolayer (SAM) on the gold working electrode, providing the specific recognition layer [3]. |
| Methylene Blue (MB)-tagged DNA | Acts as the redox reporter; its hybridization to the surface probe generates the faradaic current [3]. |
| Control DNA (without MB) | Used on the background electrode (W2) to match the chemical composition and non-faradaic properties of the sensor electrode (W1) [3]. |
| 6-Mercapto-1-hexanol (MCH) | A co-absorbate used to backfill the SAM on the gold electrode, reducing non-specific adsorption and improving hybridization efficiency [3]. |
| Gold Electrodes (paired) | The transducer surface. A matched pair is critical for analog subtraction to ensure matched capacitive backgrounds [3]. |
| Differential Potentiostat (DiffStat) | Specialized hardware for analog subtraction. Alternatively, a standard potentiostat is sufficient for digital subtraction [3]. |
Q6: What is a step-by-step protocol for benchmarking digital and analog subtraction using Square-Wave Voltammetry (SWV)?
Objective: To compare the performance of digital and analog subtraction methods for a DNA hybridization assay.
Materials: Paired gold working electrodes, reference electrode (e.g., Ag/AgCl), counter electrode, differential potentiostat (or standard potentiostat), thiolated DNA probe, MB-tagged DNA target, control DNA (no MB), MCH, and appropriate buffer.
Workflow Diagram:
Protocol Steps:
- Electrode Preparation:
- Clean the paired gold working electrodes.
- Immobilize identical thiolated DNA probe monolayers on both electrodes.
- Backfill with MCH to create a well-defined mixed SAM [3].
- Analog Subtraction (DiffStat) SWV:
- Place both electrodes in the same cell (with split reference/counter) or in identical, separate cells.
- To the first working electrode (W1), add the MB-tagged DNA target. To the second (W2), add the control DNA.
- Run SWV using the DiffStat. The potentiostat outputs the differential faradaic current in real-time [3].
- Digital Subtraction SWV:
- Use a standard single working electrode.
- In a buffer solution, run an SWV scan to serve as the background.
- Add the MB-tagged DNA target to the same cell.
- Run a second, identical SWV scan to serve as the analytical signal.
- In data analysis software, subtract the background scan from the analytical scan [46].
- Benchmarking and Analysis:
- Quantitative Comparison: Calculate and compare the signal-to-background ratio and the limit of detection (LOD) for both methods. The literature shows analog subtraction can provide order-of-magnitude improvements [3] [48].
- Qualitative Comparison: Compare the simplicity of the resulting voltammograms and the ease of data processing.
Advanced Applications & Troubleshooting
Q7: How can I perform real-time background drift correction in complex samples like serum?
This is a key strength of the analog subtraction method. The two-electrode DiffStat system can continuously correct for background drift.
- Setup: The sensor electrode (W1) is functionalized with the DNA recognition probe. The background electrode (W2) is functionalized with a non-responsive DNA sequence or is blocked with a passive SAM (e.g., MCH only).
- Measurement: Both electrodes are exposed to the flowing or static complex matrix (e.g., 50% human serum). Non-specific adsorption and matrix effects cause the background current to drift on both electrodes similarly.
- Output: The DiffStat continuously subtracts the drifting background of W2 from the combined signal+background of W1. The differential output is stable, showing only changes due to specific binding to the probe on W1 [3].
Q8: What is Continuous Square-Wave Voltammetry (cSWV) and how does it relate to digital subtraction?
cSWV is an advanced digital method that continuously collects current data throughout the entire potential pulse, unlike traditional SWV which only samples current at the end of each pulse [49].
- Relation to Digital Subtraction: The rich, continuous dataset from cSWV allows for powerful post-processing and digital subtraction. Researchers can regenerate multiple voltammograms from a single sweep, simulating different frequencies and effectively isolating faradaic information from the capacitive decay profile [49].
- Advantage: It maximizes information content from a single experiment and can be used to optimize sensing parameters rapidly without multiple physical scans.
Core Concepts: SNR, LOD, and LOQ
What are the fundamental metrics for assessing detection sensitivity?
In analytical chemistry, the Signal-to-Noise Ratio (SNR), Limit of Detection (LOD), and Limit of Quantification (LOQ) are the primary metrics for evaluating and validating the sensitivity of a method. These parameters are intrinsically linked, with SNR being the foundational element for the other two.
- Signal-to-Noise Ratio (SNR): This is a measure of how clearly an analyte can be distinguished from the background baseline noise of the analytical method [51]. It is calculated by comparing the height of the analyte signal to the average height of the baseline noise [52].
- Limit of Detection (LOD): This is the lowest concentration of an analyte that can be reliably detected, but not necessarily quantified, under the stated experimental conditions [53]. According to the ICH Q2(R1) guideline, the LOD is generally determined by an SNR between 2:1 and 3:1, with a forthcoming revision (Q2(R2)) specifying a ratio of 3:1 [51]. In practice, for robust real-world methods, an SNR of 3:1 to 10:1 is often used for LOD [51].
- Limit of Quantification (LOQ): This is the lowest concentration of an analyte that can be reliably quantified with acceptable precision and accuracy. The ICH guideline stipulates a typical SNR of 10:1 for the LOQ [51]. However, for challenging methods, a higher SNR from 10:1 to 20:1 may be required to ensure reliable quantification [51].
Table 1: Definitions and Calculation Criteria for Key Sensitivity Metrics
| Metric | Definition | Typical Signal-to-Noise (SNR) Basis | Regulatory Context (ICH Guideline) |
|---|---|---|---|
| Signal-to-Noise (SNR) | Ratio of the analyte signal height to the baseline noise height [52] | N/A | N/A |
| Limit of Detection (LOD) | The lowest concentration that can be reliably detected [53] | 3:1 to 10:1 (practical range) [51] | 3:1 (acceptable estimate per ICH Q2(R2) draft) [51] |
| Limit of Quantification (LOQ) | The lowest concentration that can be reliably quantified [51] | 10:1 to 20:1 (practical range) [51] | 10:1 (acceptable estimate) [51] |
Troubleshooting Guide: Improving SNR, LOD, and LOQ
Improving sensitivity is a systematic process of enhancing the signal and/or reducing the noise. The following guides address specific issues researchers encounter.
FAQ: How can I increase the analyte signal in my HPLC method?
A low signal is often the primary limitation. Enhancing it involves optimizing detection and chromatographic parameters.
Table 2: Strategies for Increasing Analytical Signal
| Strategy | Technical Implementation | Key Consideration |
|---|---|---|
| Optimize Detection Wavelength | For UV detection, operate at the analyte's λmax. For multiple compounds, use a compromise wavelength or multi-wavelength detection [53]. | Wavelengths below 220 nm offer strong response for many organics but may reduce selectivity [52]. |
| Improve Chromatographic Efficiency | Use columns with smaller particles (e.g., 3-μm instead of 5-μm) [52] or core-shell particles [54]. | Increases pressure; ensure system compatibility. |
| Reduce Column Volume | Switch to a column with a smaller internal diameter (e.g., 2.1 mm instead of 4.6 mm) or a shorter length [52]. | Requires proportional reduction of flow rate to maintain linear velocity. Risk of column overloading. |
| Optimize Retention Factor (k) | Adjust the mobile phase to achieve a lower k-value (e.g., 1 < k < 10) for narrower, taller peaks [52]. | Ensure the peak of interest does not co-elute with early-eluting matrix interferences. |
| Increase Injected Mass | Inject a larger volume or a more concentrated sample [52]. | Use a weak injection solvent to avoid peak distortion with large volumes [52]. |
| Leverage Alternative Detectors | For less polar compounds, switch from UV to Evaporative Light Scattering (ELSD) or use a more modern UV detector design [52]. | Detector choice depends on analyte properties and available instrumentation. |
FAQ: How can I reduce baseline noise in my HPLC-UV system?
A noisy baseline can obscure trace-level analytes. Reduction strategies range from simple electronic filtering to mobile phase optimization.
Table 3: Strategies for Reducing Baseline Noise
| Strategy | Technical Implementation | Key Consideration |
|---|---|---|
| Optimize Detector Time Constant | Increase the time constant (response/rise time) to electronically filter high-frequency noise [52]. | Set to ~1/10 the width of the narrowest peak of interest to avoid "clipping" peak tops or losing resolution [52]. |
| Adjust Data Acquisition Rate | Increase the "bunching" rate so the data system combines data points, reducing high-frequency noise in the processed chromatogram [52]. | The raw data acquisition rate should be fast; the processing rate should target ~20 points across a peak [52]. |
| Select UV-Transparent Solvents | Use acetonitrile instead of methanol or acetone, especially at low UV wavelengths (<220 nm) [53]. | Acetone has high UV absorbance and should be avoided [53]. |
| Use High-Purity Reagents | Employ HPLC-grade or LC-MS grade solvents and additives [54]. | Impurities in lower-grade reagents can significantly increase baseline noise and introduce ghost peaks. |
| Improve Temperature Stability | Operate the column in a thermostat-controlled oven and shield the instrument from drafts (e.g., from HVAC vents) [52]. | Temperature fluctuations in the detector cell can cause refractive index noise. |
FAQ: What advanced strategies can I use for LC-MS to achieve ultra-low LODs?
For mass spectrometric detection, the focus shifts to ionization efficiency and sample cleanliness.
Table 4: Advanced LC-MS Strategies for Lowering Detection Limits
| Strategy | Technical Implementation | Key Consideration |
|---|---|---|
| Improve Ionization Efficiency | Fine-tune source parameters (spray voltage, gas flows, temperatures). Use volatile mobile phase additives (e.g., formic acid, ammonium acetate) [54]. | Additives like TFA can suppress ionization in ESI-MS and should be avoided or used with caution [53]. |
| Reduce Flow Rates | Implement micro-LC or nano-LC with narrower columns (e.g., 1-2 mm ID or 75-100 μm ID) [54]. | Dramatically increases analyte concentration at the detector and improves ionization efficiency [54]. Requires specialized equipment. |
| Implement Advanced Sample Cleanup | Use Solid-Phase Extraction (SPE), Liquid-Liquid Extraction (LLE), or protein precipitation to remove matrix interferences [54]. | Reduces ion suppression and chemical noise, leading to a cleaner baseline and improved SNR. |
| Utilize Online Pre-concentration | Employ online SPE to automate sample cleanup and concentrate analytes directly within the LC flow path [54]. | Improves throughput, reduces manual handling errors, and enhances reproducibility. |
| Leverage High-Resolution MS | Use HRMS and ion mobility spectrometry (IMS) to separate analytes from isobaric interferences, effectively reducing chemical noise [54]. | Provides improved selectivity and confidence in identifying and quantifying trace-level components. |
Experimental Protocols
Protocol 1: Systematic Determination of LOD and LOQ via Signal-to-Noise
This protocol outlines the standard procedure for estimating LOD and LOQ directly from the chromatogram, as per ICH guidelines [51].
Workflow Overview
Materials & Reagents
- HPLC or LC-MS system
- Suitable analytical column
- Mobile phase solvents (HPLC grade)
- Blank matrix (e.g., pure solvent, stripped serum)
- Stock solution of the analyte of known concentration
- Chromatography Data System (CDS) software
Step-by-Step Procedure
- Analyze a Blank Sample: Inject the blank matrix and record the chromatogram. In a peak-free region of the chromatogram (typically where the analyte elutes), measure the baseline noise. The noise (N) is the difference between the maximum and minimum baseline deviation over a specified time period [51].
- Analyze a Low-Concentration Standard: Prepare and inject a standard of the analyte at a concentration expected to be near the LOD or LOQ.
- Measure the Analyte Signal: For the analyte peak in the standard chromatogram, measure the signal height (S) from the middle of the baseline to the apex of the peak [52].
- Calculate the Signal-to-Noise Ratio: Calculate the ratio using the formula: SNR = S / N [52].
- Determine LOD and LOQ:
Protocol 2: Correcting for Non-Faradaic Currents in Electrochemical Measurements
This protocol addresses the user's specific thesis context, providing a methodology inspired by recent research to isolate the Faradaic current from the total current in techniques like Cyclic Voltammetry (CV).
Workflow Overview
Materials & Reagents
- Potentiostat/Galvanostat for electrochemical measurements.
- Standard electrochemical cell (working, counter, and reference electrodes).
- Electrolyte solution of high purity.
- A pre-trained Deep Neural Network (DNN) model, such as the one described by Ha et al., inspired by speech-denoising algorithms [55].
- Computer with appropriate software (e.g., Python with TensorFlow/PyTorch) to run the DNN model.
Step-by-Step Procedure
- Data Acquisition: Perform a Cyclic Voltammetry (CV) experiment on your system. The raw output is the total current, which is the sum of the Faradaic current (from electron transfer reactions) and the Non-Faradaic current (primarily from capacitive charging of the double-layer) [55].
- Data Preprocessing: Format the total current data (current vs. potential) from the CV experiment as required for the DNN model input.
- AI-Based Discrimination: Input the total current data into the DNN model. The model is designed to learn the relationship between the total current and the underlying theoretical Faradaic current. It applies a series of weighted transformations through its fully connected layers to separate the signal from the noise (non-Faradaic contributions) [55].
- Result Extraction: The model outputs the predicted Faradaic current. Research has shown this method can predict Faradaic response from total current with a mean absolute percentage error (MAPE) of around 6.36%, and extract peak currents from experimental data with a MAPE of about 3.37% [55].
- Data Utilization: Use the purified Faradaic current signal for subsequent analysis, such as determining reaction mechanisms, calculating kinetic parameters, or estimating diffusion coefficients, free from the distortion of Non-Faradaic effects.
The Scientist's Toolkit: Key Research Reagent Solutions
Table 5: Essential Materials for Sensitivity Enhancement Experiments
| Item | Function / Application |
|---|---|
| Diamond Hydride Column | Effective for hydrophilic analytes in Aqueous Normal Phase (ANP) chromatography, often providing superior peak shape and signal intensity compared to standard reversed-phase columns [53]. |
| Solid-Phase Extraction (SPE) Kits | For selective sample clean-up to remove matrix interferences and pre-concentrate analytes, significantly reducing baseline noise and improving SNR [54]. |
| Volatile Mobile Phase Additives (e.g., Formic Acid, Ammonium Acetate) | Essential for LC-MS to enhance ionization efficiency without causing ion suppression or source contamination [53] [54]. |
| LC-MS Grade Solvents | High-purity solvents (water, acetonitrile, methanol) are critical for achieving low background noise in ultra-trace analysis [54]. |
| Sub-2 μm or Core-Shell Particle Columns | Provide enhanced chromatographic resolution and peak capacity, leading to sharper, taller peaks and improved signal [54]. |
| Pre-trained DNN Model for CV | An AI tool to discriminate Faradaic from Non-Faradaic currents in electrochemical data, enabling more accurate analysis of reaction kinetics [55]. |
For researchers developing electrochemical biosensors for clinical use, validating an assay's performance in a biologically relevant matrix like 50% human serum is a critical final step before clinical trials. Serum presents a complex, challenging environment with numerous interfering substances that can compromise an assay's accuracy and reliability. For electrochemical platforms, a primary obstacle is the non-faradaic current, a capacitive background current that can obscure the faradaic current generated by the redox reaction of the target analyte [3] [9]. This case study outlines a systematic approach, complete with troubleshooting guides and FAQs, for successfully performing this essential validation, with a specific focus on overcoming non-faradaic interferences.
Technical Deep Dive: Faradaic vs. Non-Faradaic Currents
To effectively troubleshoot, one must first understand the core interference.
- Faradaic Current: This is the analytical signal of interest. It is generated by the transfer of electrons between an electrode and a redox-active species (e.g., a methylene blue-tagged DNA probe) in solution. This current is directly proportional to the concentration of the target, following Faraday's law [9].
- Non-Faradaic (Capacitive) Current: This is the primary source of background interference. It arises from the rearrangement of ions and the charging of the electrical double-layer at the electrode-electrolyte interface. No electron transfer occurs across the interface. This current is highly susceptible to the composition of the sample matrix and can dwarf the faradaic signal, especially in complex media like serum [3] [9].
The table below summarizes the key differences.
Table 1: Characteristics of Faradaic and Non-Faradaic Currents
| Feature | Faradaic Current | Non-Faradaic Current |
|---|---|---|
| Origin | Electron transfer via redox reactions | Charging of the electrode-electrolyte interface |
| Governed by | Faraday's Law | Capacitive charging |
| Dependence on Surface Area | Proportional | Proportional |
| Impact of Serum | Can be attenuated by binding proteins | Significantly increased due to proteins and ions |
| Role in Analysis | Analytical signal | Background interference |
Experimental Protocol: Performance Validation in 50% Human Serum
The following protocol is adapted from methodologies used to validate electrochemical DNA-based sensors and immunoassays in complex matrices [3] [56].
Aim: To verify that an electrochemical biosensor maintains its sensitivity, specificity, and accuracy when analyzing targets in a 50% human serum matrix.
Materials:
- Potentiostat: Standard or, preferably, a Differential Potentiostat (DiffStat) [3].
- Electrochemical Cell: Standard three-electrode system (Working, Reference, Counter electrode).
- Working Electrodes: Gold electrodes functionalized with probe DNA (e.g., for E-DNA biosensors) or specific antibodies.
- Serum: Pooled human serum. Heat-inactivation (56°C for 30 minutes) may be considered to denature complement proteins [57].
- Analyte: Purified target (e.g., DNA sequence, protein like Carcinoembryonic Antigen).
- Buffer: Phosphate Buffered Saline (PBS), pH 7.4.
Methodology:
- Sample Preparation: Spike a known concentration of the target analyte into a solution of 50% (v/v) human serum in PBS. Prepare a negative control of 50% serum without the target.
- Baseline Measurement (in Buffer): Perform the electrochemical measurement (e.g., Square-Wave Voltammetry - SWV) of the target in a pure PBS buffer to establish the baseline signal-to-noise ratio.
- Validation Measurement (in 50% Serum): Perform the identical measurement using the 50% serum sample containing the target.
- Differential Measurement (Recommended): If using a DiffStat, use one working electrode (W1) for the serum sample with the target and a second, identical working electrode (W2) for the negative control serum. The instrument will perform real-time analog subtraction of the background [3].
- Data Analysis: Compare the signals obtained in buffer and in serum. Calculate key validation parameters such as signal suppression, recovery rate, and the limit of detection (LoD) in the serum matrix.
Diagram: Workflow for Validating Sensor Performance in 50% Serum
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Materials and Reagents for Validation
| Item | Function / Rationale | Example / Specification |
|---|---|---|
| Differential Potentiostat (DiffStat) | Hardware-based suppression of non-faradaic current via real-time analog subtraction from a blank working electrode [3]. | Custom-built or commercial DiffStat. |
| Human Serum (Pooled) | Provides a clinically relevant matrix for validation. Pooling reduces individual donor variability. | Commercially sourced, qualified for research use. |
| Functionalized Magnetic Nanoparticles | Used in homogenous assays (e.g., IMR) to reduce matrix interference and improve specificity [56]. | Antibody-functionalized Fe₃O₄ nanoparticles. |
| Redox Reporter Molecules | Generates the faradaic current. Must be stable and show reversible electrochemistry in serum. | Methylene Blue (MB), Ferrocene. |
| Stable Cell Culture Media | Serves as a base for serum dilution, supporting biomolecule stability during the assay [57]. | HybridoMed, RPMI 1640. |
| Phosphate Buffered Saline (PBS) | Provides a controlled ionic background for baseline measurements and sample dilution. | pH 7.4, 1X concentration. |
Troubleshooting Guide & FAQs
FAQ 1: Our sensor's signal is completely overwhelmed by high background noise in 50% serum. What can we do?
Answer: This is a classic symptom of dominant non-faradaic current. We recommend a two-pronged approach:
- Hardware Solution: Implement a Differential Potentiostat (DiffStat). This setup uses two working electrodes: one for your sample (W1) and an identical one with a background control (e.g., serum without target, W2). The DiffStat performs real-time analog subtraction of the capacitive current from W2, outputting a signal that is predominantly faradaic. Research shows this can suppress non-faradaic currents by an order of magnitude, making larger electrodes and higher sensitivity settings accessible [3].
- Assay Chemistry Solution: Transition to a homogenous assay format like Immunomagnetic Reduction (IMR). IMR uses antibody-functionalized magnetic nanoparticles and measures changes in AC magnetic susceptibility upon target binding. This method is less susceptible to optical and electrochemical interferences from serum components like hemoglobin and bilirubin, leading to higher specificity and a lower rate of false positives [56].
FAQ 2: How do we accurately determine the Limit of Detection (LoD) and Limit of Blank (LoB) in a complex matrix like 50% serum?
Answer: Follow established clinical laboratory guidelines such as the CLSI EP17-A protocol [56].
- Limit of Blank (LoB): Measure your negative control (50% serum without target) repeatedly (at least 60 replicates). The LoB is calculated as:
LoB = μ_blank + 1.645σ_blank, where μ and σ are the mean and standard deviation of the blank measurements. - Limit of Detection (LoD): Prepare a sample with a very low concentration of the target in 50% serum (a concentration expected to be just above the blank). Measure this low-level sample at least 60 times. The LoD is calculated as:
LoD = LoB + 1.645σ_low_level_sample.
This rigorous statistical approach ensures your reported sensitivity is reliable under clinically relevant conditions.
FAQ 3: We observe significant signal drift during measurements in serum. How can we correct for this?
Answer: Signal drift in serum is often caused by the non-specific adsorption of proteins (fouling) onto the electrode surface, which alters the interface properties over time.
- Real-time Correction: The DiffStat configuration is highly effective here. Since both working electrodes (W1 and W2) are exposed to the same serum matrix and are subject to the same fouling and drift, the differential subtraction inherently corrects for this common-mode drift, providing a stable baseline [3].
- Surface Passivation: Ensure your self-assembled monolayer (SAM) on the electrode is dense and well-ordered. Use backfilling agents (e.g., 6-mercapto-1-hexanol for DNA sensors) to minimize non-specific binding and create a protein-resistant surface.
FAQ 4: Our electrochemical recovery rates in serum are low. How can we verify if this is due to binding to serum proteins?
Answer: Low recovery often indicates that a portion of your target analyte is bound to serum proteins (e.g., albumin, alpha-1 acid glycoprotein) and is no longer accessible to your sensor's capture probe [58].
- Investigation via Equilibrium Dialysis: To confirm and quantify this, you can perform an equilibrium dialysis experiment. This involves placing your spiked serum sample on one side of a semi-permeable membrane and buffer on the other. After equilibration, measure the free (unbound) analyte concentration in the buffer chamber. The ratio of free to total analyte is the free fraction [58]. This data is crucial for accurately estimating the true active concentration your sensor is detecting.
Table 3: Troubleshooting Common Issues in 50% Serum Validation
| Problem | Potential Cause | Solution |
|---|---|---|
| High Background Noise | Dominant non-faradaic current from serum components. | Use a Differential Potentiostat (DiffStat) for hardware subtraction [3]. |
| Low Signal/Recovery | Target analyte binding to serum proteins [58]. | Determine the free fraction via equilibrium dialysis. Use a more sensitive detection method (e.g., IMR). |
| Signal Drift/Instability | Protein fouling on the electrode surface. | Use a DiffStat for drift correction. Optimize surface passivation to reduce non-specific adsorption. |
| Poor Precision | Inconsistent electrode surface or matrix effects. | Use a pooled serum source. Ensure rigorous electrode cleaning and functionalization protocols. |
FAQs: Choosing and Troubleshooting Electrochemical Techniques
FAQ 1: How do I choose between CV, SWV, and EIS for measuring electron transfer rates in my protein film study?
The optimal choice depends on the expected range of your heterogeneous electron transfer (HET) rate constant (kₕₑₜ). A comparative study investigating cytochrome c on alkanethiol-modified electrodes found that each technique has an ideal application window [59]:
- Cyclic Voltammetry (CV) is most applicable for studying interfaced proteins exhibiting kₕₑₜ of approximately 0.5 to 70 s⁻¹ [59].
- Square-Wave Voltammetry (SWV) is suitable for a broader range of kₕₑₜ from 5 to 120 s⁻¹ [59].
- Electrochemical Impedance Spectroscopy (EIS) is best for slower systems, with kₕₑₜ of about 0.5 to 5 s⁻¹ when using alkanethiols for immobilization [59]. Note that the same study reported differing kₕₑₜ values for the same system across techniques, highlighting the importance of method selection and the potential need for cross-validation [59].
FAQ 2: My electrochemical signals have a large background drift, particularly in complex media like serum. What can I do?
Consider moving beyond digital background subtraction in data processing to hardware-level solutions. A Differential Potentiostat (DiffStat) configuration can suppress non-Faradaic (capacitive) current in real-time through analog subtraction [60] [3].
- How it works: The DiffStat uses two working electrodes: an experimental electrode (W1) and a background electrode (W2). The signals are collected simultaneously and subtracted by an on-board instrumentation amplifier before measurement, effectively removing the common capacitive current [3].
- Benefits: This hardware subtraction provides order-of-magnitude improvements in sensitivity, allows the use of larger electrodes, and significantly simplifies data processing for techniques like SWV by outputting a signal that is predominantly Faradaic current [60] [3]. It has been successfully demonstrated for background drift correction in 50% human serum [3].
FAQ 3: What are the inherent strengths and weaknesses of EIS, CV, and SWV for background correction?
The core challenge is that non-Faradaic current acts as a non-zero baseline, limiting the signal-to-noise ratio and dynamic range [3]. Each technique interacts with this background differently.
- Cyclic Voltammetry (CV):
- Cons: The non-Faradaic current appears as a sloping baseline in the voltammogram, which can obscure the Faradaic peaks, especially at fast scan rates or for films with slow electron transfer [59].
- Square-Wave Voltammetry (SWV):
- Pros: This technique offers built-in digital background subtraction by its sampling mechanism. Currents are sampled at the end of the forward and reverse pulses, and the difference is plotted, which effectively suppresses capacitive currents [3] [61].
- Cons: Despite this digital subtraction, the non-Faradaic current is still present in the raw analog signal and can saturate the instrument's amplifiers, limiting the usable electrode surface area and sensitivity settings [3].
- Electrochemical Impedance Spectroscopy (EIS):
- Pros: EIS is powerful for deconvoluting different processes at the electrode interface by fitting data to equivalent circuit models. It can separate charge transfer resistance from capacitive elements like the double layer [61].
- Cons: The analysis is more complex and model-dependent. It is generally best suited for characterizing systems with relatively slow electron transfer rates [59].
Troubleshooting Guides
Problem: Low Signal-to-Noise Ratio in DNA-Based Assays
Potential Cause: Overwhelming non-Faradaic (capacitive) background currents, which are directly proportional to the electrode surface area and can mask the desired Faradaic signal from the redox reporter [3].
Solution Checklist:
- Implement Hardware Subtraction: Utilize a differential potentiostat (DiffStat) to suppress capacitive currents at the source via analog subtraction, providing a cleaner signal output [60] [3].
- Optimize Electrode Size: Reduce the working electrode surface area to decrease the absolute magnitude of the capacitive current. Be aware that this also reduces the Faradaic current [3].
- Leverage Pulse Techniques: Use SWV or DPV, which have inherent background suppression capabilities through their current sampling protocols [3] [61].
- Functionalize Electrodes for Specificity: Employ DNA monolayers (e.g., E-DNA biosensors) on gold electrodes to provide a selective sensing interface that minimizes non-specific interference [3].
Problem: Inconsistent Electron Transfer Rate Measurements
Potential Cause: Using an electrochemical technique outside its optimal range for the system under study, leading to inaccurate or method-dependent results [59].
Solution Checklist:
- Cross-Validate with a Second Technique: If you measure a kₕₑₜ of 50 s⁻¹ with CV, confirm the result using SWV, as its optimal range includes this value [59].
- Benchmark Your Protocol: Follow a standardized measurement protocol, especially for common reactions like the oxygen evolution reaction (OER). This includes careful selection of electrodes and electrolytes, control of external factors (temperature, light), and use of recommended instrument settings for CV, EIS, and other techniques [62].
- Simulate Your Data: Use theoretical simulations of the redox transition, as demonstrated in protein film studies, to verify that the obtained kinetic parameters are consistent with the experimental data across techniques [59].
Table 1: Quantitative Comparison of CV, SWV, and EIS for Kinetic Studies
| Feature | Cyclic Voltammetry (CV) | Square-Wave Voltammetry (SWV) | Electrochemical Impedance Spectroscopy (EIS) |
|---|---|---|---|
| Optimal kₕₑₜ Range [59] | 0.5 - 70 s⁻¹ | 5 - 120 s⁻¹ | 0.5 - 5 s⁻¹ |
| Example kₕₑₜ Measurement [59] | 47.8 (±2.91) s⁻¹ | 64.8 (±1.27) s⁻¹ | 26.5 s⁻¹ |
| Background Correction Method | Post-measurement digital baseline subtraction | Built-in digital sampling & hardware analog subtraction | Model-based deconvolution in equivalent circuit fitting |
| Primary Advantage | Intuitive for studying redox thermodynamics and kinetics. | High sensitivity and built-in background rejection. | Separates kinetic and capacitive interface properties. |
| Primary Disadvantage | Sloping capacitive baseline can obscure Faradaic peaks. | Raw signal still susceptible to analog saturation. | Complex data analysis; best for slower kinetics. |
Table 2: Research Reagent Solutions for Background-Corrected Assays
| Reagent / Material | Function in the Experiment |
|---|---|
| COOH-terminated Alkanethiols (e.g., C10) | Forms a self-assembled monolayer (SAM) on gold electrodes for stable, electrostatic immobilization of redox proteins like cytochrome c [59]. |
| Methylene Blue (MB)-appended DNA | Acts as a redox reporter in DNA-based monolayer sensors; its Faradaic signal is monitored against the non-Faradaic background [3]. |
| Differential Potentiostat (DiffStat) | Specialized hardware that uses two working electrodes for real-time analog subtraction of capacitive current, enhancing S/N ratio [60] [3]. |
| Multi-Electrode System (e.g., Cu, Ni, C) | Electrodes with different inherent redox properties generate complementary datasets, enriching data diversity for machine learning analysis [61]. |
Experimental Protocols
Protocol 1: Suppressing Non-Faradaic Current with a Differential Potentiostat
This protocol is adapted from studies using DNA monolayer sensors [3].
- Electrode Preparation: Fabricate two gold working electrodes (W1 and W2). Use a split reference electrode (e.g., Ag/AgCl) and counter electrode (e.g., Pt) to prevent cross-contamination between the two electrochemical cells.
- Surface Functionalization: Immobilize thiolated DNA probes on both W1 and W2 to form a self-assembled monolayer.
- Differential Setup: To W1, add the target analyte (e.g., MB-labeled complementary DNA). To W2, add a non-redox-active control sequence (CTR-DNA) to carefully match the non-Faradaic components between the two electrodes.
- Instrument Connection: Connect the electrodes to the differential potentiostat (DiffStat), with W1 as the primary working electrode and W2 as the background electrode.
- Measurement: Run the desired electrochemical technique (e.g., Chronoamperometry, CV, or SWV). The DiffStat will perform real-time analog subtraction of the background (from W2) from the analytical signal (from W1).
- Analysis: The output signal will be a current with a significantly suppressed non-Faradaic baseline, simplifying the extraction and quantification of the Faradaic component.
Protocol 2: Cross-Validating Electron Transfer Rates in Protein Films
This protocol is based on a comparative study of immobilized cytochrome c [59].
- Sample Preparation: Immobilize your redox protein (e.g., cytochrome c) electrostatically on a modified electrode surface, such as a COOH-terminated alkanethiol SAM on a silver electrode.
- Cyclic Voltammetry (CV) Measurement:
- Setup: Use a standard three-electrode potentiostat.
- Acquisition: Record CV scans at multiple scan rates.
- Analysis: Extract the kₕₑₜ (e.g., ~48 s⁻¹) from the scan rate dependence of the peak potential separation using Laviron's method or other fitting procedures.
- Square-Wave Voltammetry (SWV) Measurement:
- Setup: Use the same electrochemical cell and potentiostat.
- Acquisition: Record SWV voltammograms, optimizing frequency and amplitude.
- Analysis: Extract the kₕₑₜ (e.g., ~65 s⁻¹) by fitting the SWV net peak current versus frequency or using direct modeling.
- Electrochemical Impedance Spectroscopy (EIS) Measurement:
- Setup: Use the same electrochemical cell.
- Acquisition: Perform impedance measurements over a suitable frequency range (e.g., from 0.1 Hz to 100 kHz) at the formal potential of the protein.
- Analysis: Fit the impedance data to an appropriate equivalent circuit (e.g., a Randles circuit) to extract the charge transfer resistance (Rₜc), from which kₕₑₜ (e.g., ~27 s⁻¹) can be calculated.
- Validation: Compare the kₕₑₜ values obtained from all three methods. Use theoretical simulations of the redox transition to support the analysis and identify the most consistent and reliable value for your system.
Technical Diagrams
Decision Guide for Background Correction
DiffStat vs ConStat Signal Paths
Conclusion
Effectively correcting for non-Faradaic currents is not merely a technical exercise but a fundamental requirement for advancing the sensitivity and reliability of electrochemical biosensors in biomedical research and drug development. As synthesized from the discussed intents, a multifaceted approach—combining a deep understanding of interfacial processes, innovative hardware like the DiffStat, robust methodological choices, and rigorous validation—is key to success. These strategies collectively enable order-of-magnitude improvements in detection limits and facilitate operation in clinically relevant environments such as serum. Future directions will likely involve the deeper integration of these correction methods into point-of-care devices, the development of novel nanostructured electrode materials with intrinsic low background, and the application of these refined techniques to monitor low-abundance biomarkers and therapeutic drug levels, ultimately accelerating translational diagnostics and personalized medicine.
References
- 1. Understanding Faradaic vs Nonfaradaic Processes in ... [https://www.linkedin.com/posts/hassan-adnan-37565426b_faradaic-vs-nonfaradaic-processes-charge-activity-7367112224559063040-Kmms]
- 2. The difference between Faradaic and non ... [http://ui.adsabs.harvard.edu/abs/2018arXiv180902930B/abstract]
- 3. Non-Faradaic Current Suppression in DNA Based ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6917968/]
- 4. Intrinsic electrocatalytic activity of platinum grain boundaries [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d5sc03829d]
- 5. New electrochemical approach for assessing surface ... [https://www.sciencedirect.com/science/article/pii/S0013468625000064]
- 6. Double layer (surface science) [https://en.wikipedia.org/wiki/Double_layer_(surface_science)]
- 7. What is an Electric Double Layer? [https://interfaces.che.wisc.edu/what-is-an-electric-double-layer/]
- 8. Electric Double Layer - an overview [https://www.sciencedirect.com/topics/engineering/electric-double-layer]
- 9. Nonfaradaic Process - an overview [https://www.sciencedirect.com/topics/engineering/nonfaradaic-process]
- 10. 2: Electrical Double Layer and Charging Current [https://chem.libretexts.org/Bookshelves/Analytical_Chemistry/Supplemental_Modules_(Analytical_Chemistry)/Analytical_Sciences_Digital_Library/Courseware/Analytical_Electrochemistry%3A_The_Basic_Concepts/03_Fundamentals_of_Electrochemistry/A._Electrochemical_Thermodynamics/02_Electrical_Double_Layer_and_Charging_Current]
- 11. Faradic and non-Faradic current in Voltammetry [https://chemistry.stackexchange.com/questions/162887/faradic-and-non-faradic-current-in-voltammetry]
- 12. Non-Faradaic optoelectrodes for safe electrical ... [https://www.nature.com/articles/s41467-023-44635-8]
- 13. Sensitivity studies and optimization of an impedance- ... [https://www.sciencedirect.com/science/article/pii/S2590137024000438]
- 14. Faradaic and non-faradaic depletions by anodic electrode ... [https://www.sciencedirect.com/science/article/abs/pii/S025405842401321X]
- 15. Evaluation of Non-Faradaic Impedimetric Parameters for IL ... [https://www.mdpi.com/2072-666X/16/4/395]
- 16. Signal Amplification and Noise Reduction in ... [https://www.labx.com/resources/signal-amplification-and-noise-reduction-in-electrophysiology-data/5624]
- 17. Optimization of signal-to-noise ratio in short-duration SEP ... [https://pubmed.ncbi.nlm.nih.gov/37030046/]
- 18. Electrode Surface - an overview [https://www.sciencedirect.com/topics/engineering/electrode-surface]
- 19. What would happen if the low noise amplifier (LNA) is ... [https://ham.stackexchange.com/questions/20795/what-would-happen-if-the-low-noise-amplifier-lna-is-saturated]
- 20. The Effects of Electrode Impedance on Data Quality and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2902592/]
- 21. Reference Electrode Storage and Master ... [https://pineresearch.com/support-article/reference-electrode-storage-and-master-reference-electrodes/]
- 22. Electrochemistry in a Two- or Three-Electrode ... [https://www.mdpi.com/2072-666X/14/4/722]
- 23. Ec Faqs: Electrodes [https://www.basinc.com/manuals/EC_epsilon/EC_FAQs/faqelec]
- 24. Electrochemical essentials: Connecting reactions, currents ... [https://www.biolinscientific.com/blog/electrochemical-essentials-connecting-reactions-currents-and-potentials]
- 25. Signal-off tuned signal-on (SF-T-SN) colorimetric ... [https://www.sciencedirect.com/science/article/abs/pii/S0925400520302811]
- 26. Towards the development of a portable device based on ... [https://www.sciencedirect.com/science/article/pii/S0039914024014346]
- 27. Fluorescence Anisotropy-Based Signal-Off and ... [https://pubmed.ncbi.nlm.nih.gov/32182058/]
- 28. Signal-off impedimetric immunosensor for the detection of ... [https://www.nature.com/articles/srep19806]
- 29. Electrode pretreatment cycles for suppressing self ... [https://www.sciencedirect.com/science/article/abs/pii/S1572665725002073]
- 30. Importance of optimising electrodeposition parameters for ... [https://www.sciencedirect.com/science/article/pii/S2589234725000454]
- 31. Experimental Investigation and Optimization of the ... [https://www.mdpi.com/2079-6412/15/4/482]
- 32. MoS2/MWCNT Nanostructure: Enhanced Performance of ... [https://www.mdpi.com/2072-666X/16/4/366]
- 33. Exploring faradaic and non-faradaic electrochemical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10943381/]
- 34. Promising Solutions to Address the Non-Specific ... [https://www.mdpi.com/2227-9040/13/3/92]
- 35. New Sensors for Monitoring pH and Corrosion of ... [https://www.mdpi.com/1424-8220/22/14/5356]
- 36. All-solid-state, long term stable, and embedded pH sensor ... [https://www.sciencedirect.com/science/article/abs/pii/S2352710222009883]
- 37. Body bionics: how the next generation of biosensors could ... [https://pharmaceutical-journal.com/article/feature/body-bionics-how-the-next-generation-of-biosensors-could-revolutionise-drug-delivery]
- 38. Choosing the Right Sampling Rate for Data Acquisition [https://shop.hioki.eu/Blog/Never-Miss-a-Beat-Choosing-the-Right-Sampling-Rate-for-Data-Acquisition/?srsltid=AfmBOopKWBDxh2AjytnmXlb4oqsCVRLgnVcQC-1uUMEoG937oIv61Khe]
- 39. Optimization of Detector Parameters to Improve Sensitivity ... [https://www.waters.com/nextgen/ca/en/library/application-notes/2025/optimization-of-detector-parameters-to-improve-sensitivity-using-the-alliance-is-hplc-system-with-pda-detector.html?srsltid=AfmBOoq8notSB13MQRa9QBcsrmaXApqE7hsLJulCn0GqoWFm6qshirz0]
- 40. Optimization of Low-Cost Data Acquisition Equipment ... [https://www.mdpi.com/2227-7390/11/16/3498]
- 41. 2.4: Voltammetric and Amperometric Methods [https://chem.libretexts.org/Bookshelves/Analytical_Chemistry/Analytical_Chemistry_Volume_II_(Harvey)/02%3A_Electrochemical_Methods/2.04%3A_Voltammetric_and_Amperometric_Methods]
- 42. Antifouling zwitterionic coating enhances electrochemical ... [https://www.sciencedirect.com/science/article/pii/S1748013225002646]
- 43. Ultrarapid Method for Coating Electrochemical Sensors ... [https://pubmed.ncbi.nlm.nih.gov/34965031/]
- 44. An MOF‐Enhanced Anti‐Fouling Immunoprobe Platform for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12463068/]
- 45. Empiric Unsupervised Drifts Correction Method of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8196723/]
- 46. Background-subtraction of fast-scan cyclic staircase ... [https://www.sciencedirect.com/science/article/abs/pii/S0956566398000931]
- 47. Focusing on the role of chemometrics to enhance ... [https://www.sciencedirect.com/science/article/pii/S2214180420300441]
- 48. (Open Access) Nonfaradaic Current Suppression in DNA-Based... [https://scispace.com/papers/nonfaradaic-current-suppression-in-dna-based-electrochemical-88ow8nvyl1]
- 49. Continuous Square Wave Voltammetry for High Information Content... [https://pubmed.ncbi.nlm.nih.gov/36817008/]
- 50. Faradaic Process - an overview | ScienceDirect Topics [https://www.sciencedirect.com/topics/engineering/faradaic-process]
- 51. Why Signal-to-Noise Ratio Determines Limit of Detection [https://www.thermofisher.com/blog/analyteguru/hplc-troubleshooting-tips-why-signal-to-noise-ratio-determines/]
- 52. Enhancing Signal-to-Noise | LCGC International [https://www.chromatographyonline.com/view/enhancing-signal-noise]
- 53. How to improve LOD or detection limits in HPLC [https://www.mtc-usa.com/kb-article/aa-01024]
- 54. Advanced Strategies to Improve Detection Limits in ... [https://www.organomation.com/advanced-strategies-to-improve-detection-limits-in-chromatography-mass-spectrometry]
- 55. AI-Based Discrimination of Faradaic Current against ... [https://pubmed.ncbi.nlm.nih.gov/39754543/]
- 56. Validation of Assaying Carcinoembryonic Antigen in ... [https://www.nature.com/articles/s41598-018-28215-1]
- 57. Validation of a human-serum-based in vitro growth method for ... [https://journals.plos.org/plosntds/article?id=10.1371/journal.pntd.0009313]
- 58. Estimation of Serum-Free 50-Percent Inhibitory ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC478510/]
- 59. Cyclic voltammetry, square wave voltammetry or ... [https://www.sciencedirect.com/science/article/pii/S2667160323000248]
- 60. Nonfaradaic Current Suppression in DNA-Based Electrochemical... [https://pubmed.ncbi.nlm.nih.gov/31718147/]
- 61. Strategies to Enrich Electrochemical Sensing Data with ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11207790/]
- 62. Recommended electrochemical measurement protocol for ... [https://www.sciencedirect.com/science/article/pii/S2949881325000113]